 2019, Vol. 43
2019, Vol. 43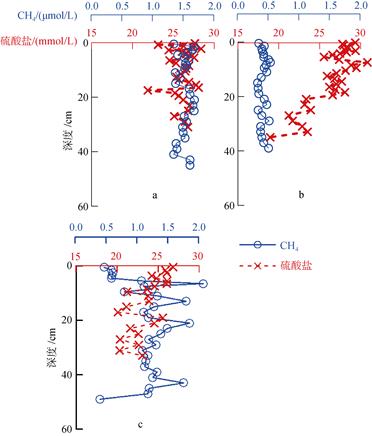



夏季东、黄海沉积物中甲烷的分布和沉积物-水界面通量

文章信息
- 叶旺旺, 谷雪霁, 康绪明, 刘素美, 张桂玲. 2019.
- YE Wang-wang, GU Xue-ji, KANG Xu-ming, LIU Su-mei, ZHANG Gui-ling. 2019.
- 夏季东、黄海沉积物中甲烷的分布和沉积物-水界面通量
- Distribution and fluxes of methane in the sediments in the East China Sea and the Yellow sea in summer
- 海洋科学, 43(7): 77-86
- Marine Sciences, 43(7): 77-86.
- http://dx.doi.org/10.11759/hykx20190326003
-
文章历史
- 收稿日期:2019-03-26
- 修回日期:2019-04-19
引用本文
叶旺旺, 谷雪霁, 康绪明, 刘素美, 张桂玲. 2019. 夏季东、黄海沉积物中甲烷的分布和沉积物-水界面通量[J]. 海洋科学, 43(7): 77-86.

YE Wang-wang, GU Xue-ji, KANG Xu-ming, LIU Su-mei, ZHANG Gui-ling. 2019. Distribution and fluxes of methane in the sediments in the East China Sea and the Yellow sea in summer[J]. Marine Sciences, 43(7): 77-86.
夏季东、黄海沉积物中甲烷的分布和沉积物-水界面通量
1. 中国海洋大学化学化工学院, 青岛 266100;
2. 海洋化学理论与工程技术教育部重点实验室, 青岛 266100;
3. 中国水产科学研究院 黄海水产研究所, 青岛 266071
2. 海洋化学理论与工程技术教育部重点实验室, 青岛 266100;
3. 中国水产科学研究院 黄海水产研究所, 青岛 266071
摘要:沉积物释放是海洋环境中甲烷(CH4)的重要来源。通过2013年7月和8月两个航次,对东、黄海泥质区沉积物中CH4浓度的垂直分布和沉积物-水界面通量进行了研究。结果表明,除个别站位外,黄海沉积物(50 cm以浅)中CH4的浓度变化范围在0.2~1.0 μmol/L之间,长江口及浙闽沿岸附近的沉积物中CH4浓度则要更高(1.0~2.0μmol/L),而东海东部海域沉积物中CH4浓度波动范围为0.2~3.0 μmol/L。总体来说,东、黄海沉积物中CH4浓度偏低,这可能与观测到的高浓度硫酸盐(>20 mmol/L)有关。通过整柱密室培养实验估算出东、黄海沉积物-水界面CH4释放速率在0.64~2.12 μmol/(m2·d)之间,东、黄海沉积物CH4释放总量为6.7×108 mol/yr;但采用菲克定律估算的CH4扩散通量则要比现场培养的结果低2~5倍,表明不同的方法在估算沉积物-水界面CH4通量上还具有一定的不确定性。
关键词:东海 沉积物 甲烷 硫酸盐 沉积物-水界面通量
Distribution and fluxes of methane in the sediments in the East China Sea and the Yellow sea in summer
1. College of Chemistry and Chemical Engineering, Ocean University of China, Qingdao 266100, China;
2. Key Laboratory of Marine Chemistry Theory and Technology Ministry of Education, Ocean University of China, Qingdao 266100, China;
3. Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, China
2. Key Laboratory of Marine Chemistry Theory and Technology Ministry of Education, Ocean University of China, Qingdao 266100, China;
3. Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, China
Abstract: Sedimentary release is an important source of marine and atmospheric CH4. of CH4 concentrations in the sediment cores in the East China Sea and the Yellow Sea were determined to investigate its vertical profiles and sediment-water CH4 fluxes. CH4 concentrations in the Yellow Sea sediments ranged from 0.2 μmol/L to 1.0 μmol/L (depth < 50 cm) except for station L06 under 40 cm (22.0 μmol/L). CH4 concentrations in the sediments in the Changjiang River Estuary and the adjacent areas ranged from 1.0 μmol/L to 2.0 μmol/L. In the eastern area of the East China Sea, CH4 concentrations in the sediments varied greatly, ranging from 0.2 μmol/L to 3.0 μmol/L. In general, low CH4 concentrations observed in the study areas may relate to the high concentration of sulfate (>20 mmol/L). We estimated the sediment -water CH4 fluxes as 0.64~2.12 μmol/(m2·d) by conducting the intact core incubation experiments. Our results combined with the previous studies indicated that the sediment-to-water CH4 emission in the East China Sea and the Yellow Sea was estimated as 6.7×108 mol/a. However, the emission rates estimated by Fick's law were 2~5 times lower than that calculated by the incubation experiments, suggesting the uncertainties when estimating the sediment-water CH4 fluxes due to different models.
Key words:
East China Sea sediment methane sulfate sediment-water flux
甲烷(CH4)作为重要的温室气体之一, 其单分子吸收红外辐射的能力比二氧化碳高20多倍。自工业革命以来, CH4在大气中的浓度从~750 ppb增长到1 803±2 ppb[1], 在全球变暖和大气化学中起着重要作用, 对温室效应的贡献可达20%[1]。海洋是大气CH4的净源, 对大气CH4的贡献仅为2%。海水中的CH4虽然只是纳摩尔级, 但是在海洋沉积物中产生的CH4要远高于此[2]。然而海洋沉积物中大部分CH4通过微生物驱动的CH4厌氧氧化过程而消耗, 在一定程度上减缓了沉积物中CH4扩散进入水体中[3]。因此CH4厌氧氧化和相伴发生的硫酸盐还原在全球CH4收支和海洋CH4向大气逸散方面有着重要的作用[4]。近年来, 河口、近海沉积物中CH4厌氧氧化和硫酸盐还原成为国际海洋研究的热点[5-6], 而国内相关研究较少, 主要集中在珠江口[7], 九龙江口[8]和南海东北部[9]沉积物中。
陆架和海湾等近岸海区虽然只占全球海洋面积的16%, 但却贡献了全球海洋CH4释放总量的75%[10]。这主要得益于较浅的水深和良好的上层通风环境, 使得沉积物中CH4能够释放到上覆水体并最终扩散到大气中[11]。东、黄海作为西北太平洋的边缘海, 其面积约为11.9×105 km2, 其中大陆架面积约占总面积的70%。海区的水文状况较复杂, 物质的来源多样, 影响了整片海区的沉积环境[12]和海水中CH4的分布。Zhang等[13-14]报道了东、黄海水体中CH4的分布规律和海-气交换通量, 并指出水体中高含量的CH4很可能来自于底部沉积物的释放。宋金明和李延[15]分析了东海沉积物间隙水中硫酸盐的分布并给出了“扩散-平流-反应”模式。Lin等[16]计算了东海陆坡沉积物中硫酸盐的还原速率。曹兴朋等[17]和Sun等[18]分别估算了春、夏季和秋、冬季东、黄海沉积物中CH4的释放通量。然而, 关于东、黄海沉积物中CH4的垂直分布还鲜有报道, 已报道的沉积物-水界面CH4交换通量也因观测站位太少而有较大的不确定性。本文首次给出了东、黄沉积物中CH4的垂直剖面, 以期探讨陆架边缘海沉积物中CH4的分布特征及其影响因素, 并利用整柱密室培养法和菲克扩散定律分别计算了CH4的沉积物-水界面交换通量, 评估了夏季该海域沉积物中CH4对水体的贡献, 并比较了两种方法获得通量的差异。
1 材料与方法 1.1 采样站位于2013年7月和8月搭乘“东方红2号”科学考察船用重力采样器(GC, 内径90 cm, 长4 m)和箱式采泥器(BC)分别在东、黄海部分海域泥质区采集了柱状沉积物, 采样站位如图 1所示。其中, L06站、F10站和P01站分别用重力采样管和箱式采泥器采集沉积物柱状样, 其他站位均用箱式采泥器采集。
 |
| 图 1 2013年7月和8月东、黄海采样站位(▽: 7月; ○: 8月; ×:培养站位) Fig. 1 Sampling locations in the Yellow Sea and the East China Sea in July (black triangles) and August (red circles), 2013. Crosses represent the stations condu cted with incubations |
在L06(114 cm)、F10(72 cm)和P01(186 cm)站利用重力采样管采集沉积物柱状样。采样后迅速用去头的5 mL注射器插入重力采样管两侧每隔6 cm预先开的小圆孔, 抽取4mL沉积物转移到盛有6 mL 1 mol/L NaOH的样品瓶中。随后用丁基橡胶塞和铝盖压紧密封, 以测定沉积物中CH4的含量。然后迅速在每个圆孔中插入土壤溶液取样器(Rhizon Soil Moisture Samplers, 荷兰), 并用真空管收集间隙水用于其他参数的分析测定。所有间隙水取完后准确转移一定体积至预先加入0.5 mL 2%醋酸锌的离心管中, 以去除硫化氢的干扰, 所有样品均分类冷冻保存。
在所有站位(包括L06、F10和P01)还用箱式采泥器(40cm×40cm)采集了沉积物, 并用有机玻璃管插管采集了沉积物柱状样(给出长度范围)。采集完毕后迅速在现场分样, 前10 cm每隔1 cm分样, 之后每隔2 cm分样, 并迅速转移到密封塑料袋中。再从不同子样品中分别按不同需要预处理样品, 处理方法和保存方法同上。
1.3 分析方法沉积物中CH4的测定采取静态顶空法[19]。其原理为通过震荡使得CH4在气相和液相之间达到平衡, 随后取2 mL顶空气体注入气相色谱仪(岛津GC-14B)进行测定。通过CH4标准气体(2.05、4.02和49.9×10–6, CH4/N2, 中国计量科学研究院)来校正仪器的信号值。沉积物CH4浓度根据沉积物的孔隙度换算为单位体积间隙水含CH4摩尔数(μmol/L)。间隙水中硫酸盐的含量采用滴定法测定[20]。沉积物孔隙度的测定采用差量称重法[21]。
1.4 沉积物-水界面CH4扩散通量 1.4.1 整柱密室培养实验在部分站位(图 1)现场开展了基于Barnes和Owens[22]方法的整柱密室培养实验以测定沉积物-水界面的CH4交换通量。选择上覆水干净且表层没有扰动的沉积物用特制的聚氯乙烯管(直径5 cm, 高度30 cm)进行插管分样。分样结束后, 取10~15根沉积柱放入培养箱内使上覆水体循环搅动并进行8~12 h的预培养以达到平衡, 然后每隔4~12 h取2~3根沉积柱终止培养, 每次取4~5个上覆水样品分别装入50 mL玻璃瓶中用于CH4和DO的测定。为了校正由于水样本身的变化或者取样时造成的培养室水相中溶解CH4浓度的变化, 在沉积物培养的同时还进行了站位底层水的培养。DO采用Winkler滴定法[23]现场测定。根据校正后的上覆水中CH4浓度随时间的变化来估算沉积物-水界面CH4通量[18]。
1.4.2 CH4扩散通量为更全面的认识不同海域的沉积物-水界面CH4通量, 本文还根据Fick第一定律计算了所有站位沉积物-水界面CH4扩散通量:
| $ J\text{=}-\phi \text{Ds}\left( \frac{\text{d}c}{\text{d}x} \right), $ | (1) |
式中J为沉积物-水界面CH4扩散通量[μmol/(m2·d)], ϕ是沉积物孔隙度, Ds是CH4在沉积物中的扩散系数, c为CH4浓度(μmol/L), x为沉积物深度(m)。扩散系数Ds根据经验公式计算[24]:
| $ \text{Ds}=\frac{{{D}_{0}}}{1+n\left( 1-\phi \right)}, $ | (2) |
其中取n=3(砂质黏土), D0是CH4在水中的扩散系数[25], t=20℃时, D0=1.75×10–5 cm2/s。
2 结果与讨论 2.1 沉积物中CH4和SO42–的垂直分布位于黄海的A02、C05和L06站位沉积物中CH4的垂直分布见图 2。A02和C05站CH4浓度在0.5 μmol/L左右波动, 并未观测到CH4浓度随深度明显增大。但L06站则表现出不同的情况, 虽然在顶部5 cm CH4浓度较低, 之后CH4浓度随深度增加明显增大, 并且在40 cm深度处浓度达到了22.0 μmol/L。这与本实验室之前在相同站位得到的结果是一致的(未发表), 推测此处可能有特殊CH4来源, 但具体情况还不清楚。L06站重力管柱状样分析结果表明, 虽然在上层60 cm沉积物中也观测到CH4浓度的明显增大, 但是该柱状样中CH4含量总体上远小于用箱式采样器在该站位获得的结果。L06站重力管与箱式采样器柱样中CH4浓度差别较大的原因, 可能是由于该站沉积物中CH4含量较高, 重力管采样时不断的震动导致沉积物中CH4气体大量溢出所致。另外, 在采样过程中, 重力管柱状样和箱式柱状样采集的时间间隔较长, 可能采样的具体区域有所差异。
 |
| 图 2 夏季黄海沉积物中CH4的分布(a为A02和C05站; b为L06站用箱式采样的分布图; c为L06站用重力管采样的分布图; BC: boxer corer, 箱式采泥器; GC: gravity corer, 重力采样器, 不标注的均为BC) Fig. 2 Depth profiles of CH4 in sediments cores from the Yellow Sea during summer (a: profiles at stations A02 and C05; b: samples at station L06 by BC; c: samples at station L06 by GC; BC: boxer corer; GC: gravity corer) |
位于长江口及浙闽沿岸的M0、O1、P01、C3、D1和E1站水深均小于30 m, 其沉积物中CH4浓度集中在1.0~2.0 μmol/L之间(图 3), 整体随深度增加而缓慢增大。其中位于长江口的O1站CH4浓度明显高于其他站位, 但硫酸盐浓度(平均值24.2 mmol/L)整体偏低, 该站底层水盐度为26.1, 沉积物组成多为粉砂、淤泥, 明显受到长江冲淡水的影响。P01站位利用重力管所取的沉积物样品最深为200 cm, CH4的含量在浅表层沉积物中比较低, 随着沉积物深度增加, CH4含量升高, 在40~60 cm深度升高明显, 并达到最高浓度2.5 μmol/L, 70 cm以下的沉积物中CH4的含量变化则趋于平稳, 平均浓度在1.9 μmol/L左右(图 3)。而在P01站利用箱式采泥器采集的沉积物样品中, CH4含量随深度有明显的升高趋势, 特别是在5~16 cm层(图 3), 但是其分布趋势与利用重力管得到的上层沉积物中的分布结果并不完全一致, 表明这两种采样方法所得结果存在一定的差异。
 |
| 图 3 长江口及浙闽沿岸附近沉积物中CH4的分布(a为M0和O1站; b为C3和D1站; c为E1和P01站; d为P01站用重力管采样的分布图) Fig. 3 Depth profiles of CH4 in sediments cores from the Changjiang Estuary and adjacent areas. (a: profiles at stations M0 and O1; b: profiles at stations C3 and D1; c: profiles at stations E1 and P01; d: samples at station P01 by GC) |
位于浙闽沿岸附近的D1和E1站沉积物中CH4平均浓度分别为1.6 μmol/L和1.4 μmol/L, 要高于长江口外的C3站(1.5 μmol/L)和长江口北部的M0站(0.5 μmol/L), 呈现出由南向北逐渐降低的趋势。整体上来说, 位于东海浙闽沿岸的D1、E1和C3站位沉积物中CH4浓度要略高于黄海A02和C05站, 表明在河口区及邻近海域丰富的有机质输入[26]会对沉积物中CH4的分布造成一定的影响, 沉积物中有机质的含量是决定沉积物中CH4向上扩散的重要因素[2]。在长江口及浙闽沿岸附近, 一方面丰富的有机质通过矿化分解加快硫酸盐的消耗。例如在东海的研究发现硫酸盐还原速率与有机碳的埋藏速率成正比[27-28], 而东海南部区域有机碳埋藏速率较高, 说明该区域内沉积物中硫酸盐的消耗较快, 浓度较低。这样较低浓度的硫酸盐可能有利于上层沉积物中CH4的产生和底部沉积物中CH4的向上扩散。另一方面丰富的有机质含量有可能促进沉积物底部产CH4过程的发生, 从而使得向上扩散的CH4浓度要高于其他地区。
A7、F10和C12位于东海东部, 比较靠近外海海区。在A7站位的垂直变化图可看出(图 4), 浅表层的沉积物中CH4的含量很低, 最低值仅为0.2 μmol/L, 之后开始随深度而升高, 在14 cm层面达到最大浓度2.7 μmol/L, 然后在14~26 cm范围内趋于平稳并在26 cm后呈现出CH4的持续降低。在10~30 cm内出现的这一段较宽的CH4峰值有可能是受到了较强的生物扰动的结果。上层沉积物中产CH4古菌有可能利用了硫酸盐还原菌不予竞争的电子受体, 例如甲醇等[7]进行产CH4活动, 从而引起沉积物特定区域内CH4含量的增加。F10(BC)和C12站的CH4含量在0.1~1.0 μmol/L之间变化, 要显著小于河口及沿岸附近的站位(1.0~2.0 μmol/L)。F4站位于东海陆架西南边, 水深73 m, 底层盐度33.9, 水温24.6℃, 有可能长期受到台湾暖流的影响, 其沉积物CH4的含量变化并不明显。总体来说这些站位沉积物中CH4含量基本处于稳定状态, 一方面表明这些站位受陆源输入的影响较少, 有机质的含量比较稳定; 另一方面说明由于水深较深, 较高活性的有机质可能在沉降过程中被微生物分解了, 而埋藏在沉积物中的主要是难降解的高分子化合物, 致使通过有机质矿化分解消耗的硫酸盐较少[2], 同时也使沉积物底部通过有机质降解的CH4较少, 低活性的有机质和高浓度的硫酸盐限制了沉积物底部CH4的含量和向上扩散的强度, 使得表层沉积物中CH4含量基本稳定。
 |
| 图 4 东海陆架泥质区沉积物中CH4的分布(a为A7和F4站; b为C12和F10站; c为F10站用重力管采样的分布图) Fig. 4 Depth profiles of CH4 in sediment cores from continental areas in the East China Sea (a: profiles at stations A7 and F4; b: profiles at stations C12 and F10; c: samples at station F10 by GC) |
在海洋沉积物中, CH4的厌氧氧化是主要的CH4消耗过程, 且通常伴随着硫酸盐的消耗[2]。图 5是黄海的C05、东海的A02和P01站沉积物中CH4和硫酸盐的垂直剖面。可以看出, 近海陆架区沉积物中CH4浓度较低, 硫酸盐浓度较高。沉积物中硫酸盐的分布主要受上覆水中向下扩散的硫酸盐速率和再矿化过程中消耗的硫酸盐速率的制约。由于采样深度所限, 未能观测到显著的CH4厌氧氧化-硫酸盐还原过程。各个站位硫酸盐和CH4的相关关系并不明显(r2 < 0.1), 可能是因为硫酸盐的浓度受再矿化过程的影响较大。但是从不同采样深度中CH4和硫酸盐的分布趋势可以看到其分布具有一定的关系, 表现为在硫酸盐浓度较高的层次, CH4相对较低; 而在硫酸盐浓度较低的层次, CH4浓度则较高(在层次上出现1~2 cm的滞后性, 可能是由于不同柱状样在分样过程中造成的层次上的误差)。一方面, 高浓度的硫酸盐可以与沉积物中CH4发生耦合作用使CH4消耗; 另一方面, 硫酸盐还原菌还可以竞争产CH4基质从而限制CH4产生。因此, 在浅表沉积物中硫酸盐含量对CH4分布特征具有重要影响。
于C05、A7和D12站位进行了沉积物整柱密室培养实验, 结果如图 6所示。随着时间的推移, 培养柱上覆水中CH4浓度有了明显的增长。D12站位于东海东南边, 临近冲绳海槽, 水深780 m。其沉积物培养实验表明, 在前20小时, CH4增长缓慢, 到56小时, 有了迅速的增加。同样的情况发生在A7站, 即在0-48小时内, CH4浓度有了明显的增长, 并且伴随DO消耗的同时在48小时达到了最高值。C05站DO的消耗速度比A7站更快, 在培养结束后达到低值, 同时CH4浓度也达到了最高值。根据实验结果和校正后的CH4浓度, 估算出D12站沉积物-水界面CH4释放通量为0.64 μmol/(m2·d), 而A7站和C05站分别为1.30 μmol/(m2·d)和2.12 μmol/(m2·d), 这与之前在相同海域相同季节观测到的结果类似(1.05- 5.52 μmol/(m2·d))[18], 表明东、黄海夏季沉积物是水体CH4的源, 并且不同区域之间沉积物CH4释放量有明显的不同, 表现出较大的空间差异性。
 |
| 图 6 CH4(nmol/L)和DO(mg/L)在D12(a)、A7(b)和C05(c)站沉积物培养实验中的变化及沉积物-水界面CH4扩散通量(d) Fig. 6 CH4 (nmol/L) production and DO (mg/L) consumption in the incubation experiments. (a) D12, (b) A7, (c) C05; (d) sediment-water CH4 fluxes at those stations |
表 1汇总了文献报道的不同环境沉积物-水界面CH4扩散通量, 结果显示, 不同区域沉积物释放的速率有较大的不同。在海底极端环境中CH4的扩散通量要比陆架边缘海高几个数量级[29], 如冷泉渗漏区[30]沉积物释放通量可达1.37×105 μmol/(m2·d)。而河口海湾区的变化范围则较大(0.1~8 000 μmol/(m2·d)), 其沉积物中CH4扩散通量也要比陆架边缘海高很多[31]。Segarra等[31]研究表明乔治亚州海岸沉积物中CH4的产生和释放在冬季处于一个低值, 这可能与不同季节温度的变化[32]、CH4的溶解度[33]和初级生产力[34]有关。从表 1也可以看出, 不同模型和计算方法对沉积物-水界面CH4的释放通量会有一定的影响。东、黄海属于西北太平洋的陆架海, 既受到河流陆源输入的影响, 也受到外海黑潮水入侵的影响, 所以其有机质来源有较大不同。而沉积物培养站位都处于陆架外部与外海相连的地方, Lin等[16]发现东海南部陆坡沉积物有机碳埋藏速率较高, 但是对应的硫酸盐浓度并不高。他们认为陆架陆坡处沉降的有机物经过长距离的输送会使容易降解的有机质先分解, 而难于降解的有机质则埋藏于沉积物底部。这些活性不高的有机质则会影响到沉积物底部CH4的扩散速率, 因此其沉积物-水界面CH4扩散通量会处于一个比较低的范围。
表 1 不同区域沉积物-水界面CH4扩散通量 Tab. 1 Sediment-water CH4 fluxes in different areas
| 调查区域 | 类型 | 时间 | 通量/[μmol/(m2·d)] | 计算方法 | 参考文献 |
| 孟加拉湾东北海岸 | 红树林 | 2003.12 | 1.97~134.6 | 现场测定 | [30] |
| 卡斯卡底古陆边缘 | 冷泉渗漏区 | 1999.7 | 1.37×105 | 数值模型 | [35] |
| 黑海水槽 | 泥火山 | 2007.2—3 | 4.6×105 | 密室培养 | [36] |
| 乔治亚州海岸 | 淡水河流 | 2008—2009 | (0.1~55.8)×103 | 密室培养 | [31] |
| 东地中海 | 冷泉渗漏区 | 2006, 2007, 2009 | (36~881)×103 | 密室培养 | [29] |
| 西班牙南海岸 | 河流 | 2013 | 1.2~8300 | 菲克定律 | [37] |
| 珠江口 | 河口 | 2004.11—12; 2006.3 | 0.1~0.4 | 菲克定律 | [7] |
| 南海 | 陆架边缘 | 0.03~23.8 | 菲克定律 | [38] | |
| 东、黄海 | 河口陆架 | 2002.4—5 | 1.73~2.21 | 密室培养法 | [14] |
| 2011.3 | –0.08~1.67 | 密室培养法 | [18] | ||
| 2011.5 | 0.72~2.51 | 密室培养法 | [18] | ||
| 2011.8 | 1.05~5.52 | 密室培养法 | [18] | ||
| 2011.10 | 0.54~0.94 | 密室培养法 | [18] | ||
| 2011.12 | 0.38~0.72 | 密室培养法 | [18] | ||
| 2013.7—8 | 0.64~2.12 | 密室培养法 | 本研究 | ||
| 2013.7—8 | 0.03~1.84 | 菲克定律 | 本研究 |
将本文结果的平均值[1.35 μmol/(m2·d)]和以往东、黄海沉积物CH4扩散速率以及东、黄海海域面积(11.9×105 km2)可以估算出东、黄海沉积物CH4释放量为6.7×108 mol/a。当然这种初步的估算没有考虑到区域性的变化所来带的误差。从表 1也可以看出, 不同模型和计算方法对沉积物-水界面CH4的释放通量会有一定的影响(见2.2.2节)。
2.2.2 菲克定律计算沉积物-水界面CH4扩散通量利用菲克第一定律估算得到各个站位沉积物中CH4扩散通量范围为0.03~1.84 μmol/(m2·d), 平均值为0.68±0.68 μmol/(m2·d), 其中位于河口区的O1站CH4扩散通量最高, 其次为黄海的L06站和长江口附近的P01站(图 7), 这表明陆源输入对沉积物CH4释放有明显影响。扩散速率最小的站是黄海南部的M0站和东海西部的C3站。这些站位较低的扩散通量可能是由于水深较浅, 受水动力因素的影响较大所致。特别是C3站可能在采样过程中受到的扰动较大。其他站位也表现出不同的CH4扩散速率, 表明沉积物中CH4的扩散通量具有较大的空间差异性。
 |
| 图 7 不同站位沉积物-水界面CH4扩散通量 Fig. 7 Sediment-to-water CH4 fluxes at different stations |
与整柱密室培养法计算得到的CH4扩散通量相比, 菲克第一定律计算的结果低2~5倍(A7= 0.66 μmol/(m2·d)和C05=0.38 μmol/(m2·d), 图 7)。这是因为菲克定律计算的CH4扩散通量只考虑了沉积物中CH4扩散的物理过程, 并且利用沉积物中不同层次的CH4浓度的差值来估算的。受采样条件的限制, 我们的沉积物表层只取到1 cm的地方且分样过程时间较长, 即使是在沉积物4~5 cm的深度, 其不确定因素也有很多, 因此这种近似的估算忽略了沉积物和沉积物-水界面中生物化学过程的影响, 如CH4的产生等。更重要的是, 在取样和分样过程中不可避免的会对沉积物造成扰动, 从而引起沉积物中CH4与大气的交换, 进而降低估算的结果。所以基于菲克定律计算的速率仅代表了CH4在沉积物中的扩散速率并且还有相当大的不确定性。
由于实验方法的局限和现场条件的限制, 本文采用的整柱密室培养法也存在一定的误差。例如采样过程中对扰动小的沉积物的把握和培养过程中温度的控制等。虽然尽量避免人为因素对其培养过程的影响, 但是培养过程中生物扰动等未知因素也会使结果产生一定的偏差。此外, 由于培养是在沉积物-水界面重新达到平衡后开始, 因此经过之前的采样处理过程以及预培养环节, 并不能完全复原现场的环境, 最后可能预测的是重新平衡后的潜在结果, 会让实验结果更大的偏离实际值。因此要在以后的研究中获得更准确的数据和更真实的结果, 需要改进实验方法, 同时提高操作可行性, 减少各方面的干扰。综合考虑, 通过现场整柱密室培养估算的结果更加接近真实值。
3 结论本文通过两个航次的观测数据, 总结了东、黄海不同泥质区沉积物中CH4和硫酸盐的垂直分布规律, 结果表明河口区及浙闽沿岸附近的沉积物中CH4浓度较高, 而陆架及外海附近沉积物中CH4浓度较低。夏季东、黄海沉积物表现为水体CH4的源, 基于培养实验估算其向水体释放CH4的速率为6.7×108 mol/a。沉积物底部向上扩散的高浓度CH4有可能在沉积物浅表层被大量氧化, 有效的阻止沉积物中大量CH4进入上覆水体。基于不同方法和模型估算的沉积物-水界面通量会有较大的差异。例如, 通过菲克定律计算的沉积物-水界面CH4通量由于没有考虑存在的生物化学过程可能会处于较低的水平, 其估算的结果要比整柱密室培养实验的结果低2~5倍, 而通过现场整柱密室培养估算的结果更加接近真实值。
参考文献
| [1] |
IPCC. Climate Change 2013:the physical science basis[M]. New York: Cambridge University Press, 2013.
|
| [2] |
Reeburgh W S. Oceanic Methane Biogeochemistry[J]. Chemical Reviews, 2007, 107: 486-513. DOI:10.1021/cr050362v |
| [3] |
Hinrichs K U, Boetius A. The anaerobic oxidation of methane:new insights in microbial ecology and biogeochemistry[J]. Ocean Margin Systems, 2003, 457-477. |
| [4] |
Parkes R J, Cragg B A, Banning N, et al. Biogeochemistry and biodiversity of methane cycling in subsurface marine sediments (Skagerrak, Denmark)[J]. Environmental Microbiology, 2007, 9: 1146-1161. DOI:10.1111/j.1462-2920.2006.01237.x |
| [5] |
Pimenov N V, Kanapatskii T A, Sigalevich P A, et al. Sulfate reduction, methanogenesis, and methane oxidation in the Holocene sediments of the Vyborg Bay, Baltic Sea[J]. Microbiology, 2012, 81: 79-89. DOI:10.1134/S0026261712010122 |
| [6] |
Yoshinage M Y, Holler T, Goldhammer T, et al. Carbon isotope equilibration during sulphate-limited anaerobic oxidation of methane[J]. Nature Geoscience, 2014, 7: 190-194. DOI:10.1038/ngeo2069 |
| [7] |
吴自军, 周怀阳, 彭晓彤. 珠江口及其邻近海域沉积物甲烷-硫酸根界面分布深度及影响因素[J]. 海洋与湖沼, 2009, 40(3): 249-260. Wu Zijun, Zhou Huaiyang, Peng Xiaotong. Depth of sulfate-methane interface (SMI) in sediment and affecting factors in the Pearl River estuary and vicinal South China Sea[J]. Oceanologia et Limnologia Sinica, 2009, 40(3): 249-260. DOI:10.3321/j.issn:0029-814X.2009.03.001 |
| [8] |
尹希杰, 陈坚, 郭莹莹, 等. 九龙江河口沉积物中硫酸盐还原与甲烷厌氧氧化:同位素地球化学证据[J]. 海洋学报, 2011, 33(4): 121-128. Yin Xijie, Chen Jian, Guo Yingying, et al. Sulfate reduction and methane anaerobic oxidation:isotope geochemical evidence from the pore water of coastal sediments in the Jiulong Estuary[J]. Acta Oceanologica Sinica, 2011, 33(4): 121-128. |
| [9] |
陆红锋, 刘坚, 陈芳, 等. 南海东北部硫酸盐还原-甲烷厌氧氧化界面——海底强烈甲烷渗溢的记录[J]. 海洋地质与第四纪地质, 2012, 32(1): 93-98. Lu Hongfeng, Liu Jian, Chen Fang, et al. Shallow sulfate-methane interface in northeastern South China Sea:An indicator of strong methane seepage on seafloor[J]. Marine Geology & Quaternary Geology, 2012, 32(1): 93-98. |
| [10] |
Bange H W, Bartell U H, Rapsomanikis S, et al. Methane in the baltic and north seas and a reassessment of the marine emissions of methane[J]. Global Biogeochemical Cycles, 1994, 8: 465-480. DOI:10.1029/94GB02181 |
| [11] |
Borges A V, Speeckaert G, Champenois W, et al. Productivity and temperature as drivers of seasonal and spatial variations of dissolved methane in the Southern Bight of the North Sea[J]. Ecosystems, 2018, 21: 583-599. DOI:10.1007/s10021-017-0171-7 |
| [12] |
王伟, 李安春, 徐方建, 等. 北黄海表层沉积物粒度分布特征及其沉积环境分析[J]. 海洋与湖沼, 2009, 40(5): 525-531. Wang Wei, Li Anchun, Xu Fangjian, et al. Distribution of surface sediments and sedimentary environment in the North Yellow Sea[J]. Oceanologia et Limnologia Sinica, 2009, 40(5): 525-531. DOI:10.3321/j.issn:0029-814X.2009.05.001 |
| [13] |
Zhang G, Zhang J, Kang Y, et al. Distributions and fluxes of methane in the East China Sea and the Yellow Sea in spring[J]. Journal of Geophysical Research:Oceans, 2004, 109: C07011. |
| [14] |
Zhang G, Zhang J, Ren J., et al. Distributions and sea-to-air fluxes of methane and nitrous oxide in the North East China Sea in summer[J]. Marine Chemistry, 2008, 110: 42-55. DOI:10.1016/j.marchem.2008.02.005 |
| [15] |
宋金明, 李延. 东海沉积物间隙水中硅酸盐和硫酸盐"扩散-平流-反应"模式的研究[J]. 海洋科学, 1991, 1: 27-37. Song Jinming, Li Yan. Studies on models of "diffusion-advection-reaction" of silicate and sulfate in sediment interstitial waters of the East China Sea[J]. Marine Sciences, 1991, 1: 27-37. |
| [16] |
Lin S, Hsieh I J, Huang K M, et al. Influence of the Yangtze River and grain size on the spatial variations of heavy metals and organic carbon in the East China Sea continental shelf sediments[J]. Chemical Geology, 2002, 182: 377-394. DOI:10.1016/S0009-2541(01)00331-X |
| [17] |
曹兴朋, 张桂玲, 马啸, 等. 春季东、黄海溶解甲烷的分布和海气交换通量[J]. 环境科学, 2013, 34(7): 2565-2573. Cao Xingpeng, Zhang Guiling, Ma Xiao, et al. Distribution and air-sea fluxes of methane in the Yellow Sea and the East China Sea in the spring[J]. Environmetal Science, 2013, 34(7): 2565-2573. |
| [18] |
Sun M S, Zhang G, Ma X, et al. Dissolved methane in the East China Sea:Distribution, seasonal variation and emission[J]. Marine Chemistry, 2018, 202: 12-26. DOI:10.1016/j.marchem.2018.03.001 |
| [19] |
Treude T, Niggemann J, Kallmeyer J, et al. Anaerobic oxidation of methane and sulfate reduction along the Chilean continental margin[J]. Geochimica et Cosmochimica Acta, 2005, 69(11): 2767-2779. DOI:10.1016/j.gca.2005.01.002 |
| [20] |
Hoioarth R W. A rapid and precise method for determining sulfate seawater, estuarine waters, and sediment pore waters[J]. Limnology and Oceanography, 1978, 23: 1066-1069. DOI:10.4319/lo.1978.23.5.1066 |
| [21] |
Kang X, Liu S, Ning X. Reduced inorganic sulfur in sediments of the mariculture region of Sanggou Bay, China[J]. Aquaculture Environment Interactions, 2016, 8: 233-246. DOI:10.3354/aei00154 |
| [22] |
Barnes J, Owens N. Denitrification and nitrous oxide concentrations in the Humber estuary, UK, and adjacent coastal zones[J]. Marine Pollution Bulletin, 1999, 37: 247-260. DOI:10.1016/S0025-326X(99)00079-X |
| [23] |
Bryan J R, Rlley J P, Williams P J L. A winkler procedure for making precise measurements of oxygen concentration for productivity and related studies[J]. Journal of Experimental Marine Biology and Ecology, 1976, 21: 191-197. DOI:10.1016/0022-0981(76)90114-3 |
| [24] |
Iversen N, J rgensen B B. Diffusion coefficients of sulfate and methane in marine sediments:Influence of porosity[J]. Geochimica et Cosmochimica Acta, 1993, 57: 571-578. DOI:10.1016/0016-7037(93)90368-7 |
| [25] |
Broecker W S, Peng T H. Gas exchange rates between air and sea[J]. Tellus, 1974, 26: 21-35. |
| [26] |
Gao S, Wang Y P. Changes in material fluxes from the Changjiang River and their implications on the adjoining continental shelf ecosystem[J]. Continental Shelf Research, 2008, 28: 1490-1500. DOI:10.1016/j.csr.2007.02.010 |
| [27] |
陈建芳, 金海燕, 刘小涯, 等. 黄海和东海沉积物有机质活性及营养盐再生潜力初探[J]. 地球化学, 2005, 34(4): 387-394. Chen Jianfang, Jin Haiyan, Liu Xiaoya, et al. Reactivity and potential regenerating capability of sedimentary organic matter in the Yellow Sea and the East China Sea[J]. Geochimica, 2005, 34(4): 387-394. DOI:10.3321/j.issn:0379-1726.2005.04.010 |
| [28] |
Lin S, Huan K M, Chen S K. Organic carbon deposition and its control on iron sulfide formation of the southern East China Sea continental shelf sediments[J]. Continental Shelf Research, 2000, 20: 619-635. DOI:10.1016/S0278-4343(99)00088-6 |
| [29] |
R mer M, Sahlin H, Pape T, et al. Methane fluxes and carbonate deposits at a cold seep area of the Central Nile Deep Sea Fan, Eastern Mediterranean Sea[J]. Marine Geology, 2014, 347: 27-42. DOI:10.1016/j.margeo.2013.10.011 |
| [30] |
Biswas H, Mukhopadhyay S, Sen S, et al. Spatial and temporal patterns of methane dynamics in the tropical mangrove dominated estuary, NE coast of Bay of Bengal, India[J]. Journal of Marine Systems, 2007, 68: 55-64. DOI:10.1016/j.jmarsys.2006.11.001 |
| [31] |
Segarra K E A, Samarkin V, King E, et al. Seasonal variations of methane fluxes from an unvegetated tidal freshwater mudflat (Hammersmith Creek, GA)[J]. Biogeochemistry, 2013, 115: 349-361. DOI:10.1007/s10533-013-9840-6 |
| [32] |
Van Der Nat F J, Middelburg J J. Methane emission from tidal freshwater marshes[J]. Biogeochemistry, 2000, 49: 103-121. DOI:10.1023/A:1006333225100 |
| [33] |
Chanton J P, Martens C S, Kelley C A. Gas transport from methane-saturated, tidal freshwater and wetland sediments[J]. Limnology and Oceanography, 1989, 34: 807-819. DOI:10.4319/lo.1989.34.5.0807 |
| [34] |
Whiting G, Chanton J. Primary production control of methane emission from wetlands[J]. Nature, 1993, 364: 794-795. DOI:10.1038/364794a0 |
| [35] |
Luff R, Wallmannm K. Fluid flow, methane fluxes, carbonate precipitation and biogeochemical turnover in gas hydrate-bearing sediments at Hydrate Ridge, Cascadia Margin:Numerical modeling and mass balances[J]. Geochimica et Cosmochimica Acta, 2003, 67(18): 3403-3421. DOI:10.1016/S0016-7037(03)00127-3 |
| [36] |
Lichtschlag A, Felden J, Wenzh fer F, et al. Methane and sulfide fluxes in permanent anoxia:in situ studies at the Dvurechenskii mud volcano (Sorokin Trough, Black Sea)[J]. Geochimica et Cosmochimica Acta, 2010, 74(17): 5002-5018. DOI:10.1016/j.gca.2010.05.031 |
| [37] |
Burgos M, Sierra A, Ortega T, et al. Anthropogenic effects on greenhouse gas (CH4 and N2O) emissions in the Guadalete River Estuary (SW Spain)[J]. Science of The Total Environment, 2015, 503-504: 179-189. DOI:10.1016/j.scitotenv.2014.06.038 |
| [38] |
Lin S, Hsieh W, Lim Y C. Methane migration and its influence on sulfate reduction in the good weather ridge region, South China Sea continental margin sediments[J]. Terrestrial Atmospheric and Oceanic Sciences, 2006, 17(4): 883-902. DOI:10.3319/TAO.2006.17.4.883(GH) |