 2016, Vol. 47
2016, Vol. 47中国海洋湖沼学会主办。
文章信息
- 窦妍, 赵晓伟, 丁君, 何鹏. 2016.
- DOU Yan, ZHAO Xiao-Wei, DING Jun, HE Peng. 2016.
- 应用高通量测序技术分析北方刺参养殖池塘环境菌群结构
- APPLICATION OF HIGH-THROUGHPUT SEQUENCING FOR ANALYZING BACTERIAL COMMUNITIES IN EARTHEN PONDS OF SEA CUCUMBER AQUACULTURE IN NORTHERN CHINA
- 海洋与湖沼, 47(1): 122-129
- Oceanologia et Limnologia Sinica, 47(1): 122-129.
- http://dx.doi.org/10.11693/hyhz20150300088
-
文章历史
- 收稿日期: 2015-03-24
- 收修改稿日期: 2015-06-15




刺参(Apostichopus japonicus)是一种经济价值很高的海珍品,具有很高的药用价值和营养价值(Bordbar et al,2011; Purcell et al,2012)。国内外海鲜需求的日益增长刺激着中国海水养殖业的快速发展,然而,过去十年间养殖海洋动物疾病的频繁暴发,给养殖业造成毁灭性的失败和巨大的经济损失。细菌通常是传染病的病原体,如在刺参夏眠和户外养殖过程中暴发的腐皮综合症和细菌性溃疡综合症(Wang et al,2005),疾病的频繁暴发已成为阻碍刺参养殖业迅猛发展的一个重要原因(马悦欣等,2006; Deng et al,2009; Li et al,2010a)。水产养殖环境中细菌群落组成不仅对养殖动物内部菌群有很重要的影响,而且对养殖动物的营养、免疫和抵抗疾病也有重要的作用(Paerl et al,2003; Luo et al,2006)。因此,通过研究刺参养殖环境菌群的结构特点,可以预防细菌性疾病的暴发,对刺参健康养殖具有重要意义。
许多微生物在自然条件下处于共生体系,很难利用传统的培养方法获得纯培养物(Amann et al,1995; Hugenholtz et al,2009),且分离富集培养方法具有很强的选择性,因而该方法不能准确地反映自然状态下微生物群落的真实情况(Rosselló-Mora et al,2001; Auguet et al,2009),这就成为研究刺参菌群结构的一个难题。近几年,随着基于16S rDNA扩增子测序的分子生物学的发展,尤其是高通量测序技术的研发及应用,为微生物多样性研究策略注入了新的力量。16S rRNA扩增子测序通常是选择某个或某几个变异区域,利用保守区设计通用引物进行PCR扩增,然后对高变区进行测序分析和菌种鉴定,成为研究环境样品中微生物组成结构的重要手段(Youssef et al,2009; Caporaso et al,2011)。目前关于刺参养殖环境中菌群结构的研究已有报道,如关晓燕等(2010)、王轶南等(2010)、李建光等(2014)都对刺参养殖环境菌群结构进行过报道,但其主要研究方法为DGGE技术。DGGE指纹图谱技术,在其实验结果中往往只含有数十条条带,仅能反映出样品中少数优势菌的信息; 且由于分辨率的误差,部分电泳条带中可能包含不只一种16S rRNA序列,因此要了解电泳图谱中具体的菌种信息,还需对每一条带构建克隆文库并筛选克隆子进行测序,此实验操作相对繁琐。下一代高通量测序直接从环境样本中扩增16S rRNA序列高变区并进行测序,能同时对样品中的优势种群及微量菌进行检测,获得样品中的微生物群落组成,并将其含量进行数字化分析,因而被广泛应用于环境样品菌群多样性的研究(Bell et al,2013; Ren et al,2015; Yang et al,2015)。
辽宁省是我国刺参主要养殖区域之一,本研究采用基于Miseq平台的高通量测序技术对大连地区(普兰店、庄河、旅顺)三个养殖公司的刺参养殖环境(养殖水和沉积物)菌群结构进行了分析,以期更客观、全面地反映出刺参养殖环境菌群的真实情况,为刺参健康养殖、疾病防控提供理论依据。
1 材料与方法 1.1 样品采集
2014年10月—11月分别对大连地区(普兰店、庄河、旅顺)的三个刺参养殖公司进行样品采集(表 1)。用无菌瓶分别取刺参室内和室外养殖环境水面40cm左右处海水1L,室外刺参养殖环境底部2—5cm处底泥100g(王轶南等,2010),采集样品于冰盒中运送至实验室。
| 养殖公司 | 室内养殖池塘水样 | 室外养殖环境水样 | 室外养殖环境沉积物样 |
| 普兰店养殖公司 | DJ1 | DJ2 | DJ9 |
| 庄河养殖公司 | DJ5 | DJ6 | DJ11 |
| 旅顺养殖公司 | DJ12 | DJ13 | DJ14 |
用0.22μm无菌滤膜对水样进行过滤。水样和底泥样DNA分别用OMEGA水样DNA提取试剂盒(OMEGA Water DNA Kit,D5525)和土壤DNA提取试剂盒(OMEGA Soil DNA Kit,D5625)提取。16S rRNA基因V3-V4区扩增引物为: 341F(5′-CCTACGGGNG GCWGCAG-3′)和805R(5′-GACTACHVGGGTATCTA ATCC-3′)。送生工生物工程(上海)股份有限公司进行基于Illumina Miseq测序平台的高通量测序。
1.3 数据处理测序完成后,对原始数据进行处理,处理过程如下:
质量控制:(1)采用Prinseq软件(PRINSEQ-lite 0.19.5)对所得测序序列的3’端进行质控,截掉质量低的数据;(2)通过Flash软件(FLASH v1.2.7)融合双末端序列,并根据barcode序列将序列拆分,各自回归到对应样品;(3)采用Prinseq软件(PRINSEQ-lite 0.19.5)对各个样品进行去引物序列、短片段、低复杂度序列、低质量序列。
去除嵌合体及非靶区域序列:(1)采用Mothur软件包(Schloss et al,2009)中的Pre.cluster软件然间进行测序错误校正,校正过程当中允许的最大错配为1/150;(2)以SILVA数据库中序列作为模板,采用UCHIME(Edgar et al,2011)软件,去除数据中的嵌合体。
1.4 多样性和群落结构分析采用UCLUST软件(UCLUST v1.1.579)将序列按照序列相似性为97%的阈值进行OTUs操作分类单元(Operational Taxonomic Units,OTUs)聚类。采用Mothur软件对样品序列进行Alpha多样性分析,包括丰富度指数(Richness)、ACE指数(ACE Index)、Chao1指数(Chaos1 Index)、覆盖率(Coverage)。选择OTUs操作单元里的一条代表序列(默认丰度最高)采用RDP classifier软件进行物种分类,分类阈值默认为0.8。
2 结果与分析 2.1 测序序列处理结果9个样品所得原始测序序列为28977—42115条,原始序列上传至NCBI的SRA数据库,登录号为SRP052819。原始序列经质量控制、嵌合体和非靶区域序列去除后,9个样品所得有效序列为26503— 37825条(表 2)。
| 样品号 | 原始序列 | 质控后序列 | 非靶区序列序列 | 嵌合体序列 | 有效序列 |
| DJ1 | 41340 | 41334 | 12 | 4441 | 36881 |
| DJ2 | 40343 | 40336 | 4 | 2507 | 37825 |
| DJ9 | 37490 | 37487 | 17 | 1826 | 35644 |
| DJ5 | 38006 | 38003 | 0 | 4647 | 33356 |
| DJ6 | 42115 | 42111 | 1 | 5112 | 36998 |
| DJ11 | 38453 | 38453 | 2 | 7664 | 30787 |
| DJ12 | 28977 | 28971 | 4 | 2464 | 26503 |
| DJ13 | 30469 | 30465 | 33 | 2938 | 27494 |
| DJ14 | 29102 | 29098 | 0 | 2082 | 27016 |
本研究9个样品的有效序列经进一步分析可归为1502—5655个OTUs(图 1)。9个样品中ACE和Chao 1多样性指数分别是5579—25014和3240— 15231,各样品菌群多样性较高。除DJ9、DJ11、DJ13、DJ14样品外,样品DJ1、DJ2、DJ5、DJ6、DJ12的稀疏曲线趋于饱和(图 2)。测序样品的覆盖率为86%— 97%,其中DJ9、DJ11、DJ13、DJ14样品覆盖率值较其它样品的覆盖率值低。由多样性指数和稀疏曲线分析,DJ9样品中的菌群多样性大于DJ1和DJ2,DJ11样品中的菌群多样性大于DJ5和DJ6,DJ14样品中的菌群多样性大于DJ12和DJ13,即各公司刺参养殖环境沉积物中菌群多样性大于其相应公司的刺参养殖水体中菌群多样性。在室外养殖环境水中DJ13为未经处理的自然海域中的海水,DJ2和DJ6海水经沉淀和砂滤处理,因此DJ13中菌群多样性大于DJ6和DJ2。
 |
| 图 1 基于OUT的ACE和Chao 1多样性指数的Alpha多样性分析 Fig. 1 The Alpha diversity of bacteria associated with samples, and the numbers of OTUs observed and predicted (ACE and Chao 1) |
 |
| 图 2 样品丰富度稀疏分析图 Fig. 2 Richness rarefaction plot from all samples |
根据分类结果,9个样品中检测到的细菌归属于26个门类、69个纲、105个目、249个科、890个属,主要门类为变形菌门Proteobacteria、拟杆菌门Bacteroidetes、绿弯菌门 Chloroflexi、浮霉菌门Planctomycetes、蓝细菌门Cyanobacteria、酸杆菌门Acidobacteria、疣微菌门Verrucomicrobia、厚壁菌门Firmicutes和放线菌门Actinobacteria等(图 3),另外17个门类的细菌在9个样品中的检测量均不足1%,主要为嗜热丝菌门Caldiserica、古细菌Archaea、柔膜菌门Tenericutes、纤维杆菌门Fibrobacteres、互养菌门Synergistetes、硝化螺旋菌门Nitrospira、迷踪菌门Elusimicrobia、装甲菌门Armatimonadetes、衣原体门Chlamydiae、异常球菌-栖热菌门Deinococcus- Thermus、绿菌门Chlorobi、芽单胞菌门Gemmatimonadetes、梭杆菌门Fusobacteria、黏胶球形菌门Lentisphaerae、产水菌门Aquificae、脱铁杆菌门Deferribacteres和螺旋体门Spirochaetes等。DJ1样品中含有19个门类的细菌,其中优势菌群为拟杆菌门Bacteroidetes(52.03%),次优势菌群为变形菌门Proteobacteria(41.93%); DJ2样品中含有18个门类的菌群,其优势菌群为变形菌门(63.90%),次优势菌群为拟杆菌门(24.50%); DJ9样品中含有的细菌门类最多(21个门类),优势菌群为变形菌门(58.05%),次优势菌群为拟杆菌门(12.20%); DJ5样品中含有的细菌门类最少(12个门类),优势菌群为变形菌门(88.21%),次优势菌群为拟杆菌门(10.89%); DJ6样品中含有15个门类的细菌,优势菌群为变形菌门(50.80%),次优势菌群为拟杆菌门(38.34%); DJ11样品中含有20个门类的细菌,优势菌群为变形菌门(59.92%),次优势菌群为拟杆菌门(18.92%); DJ12样品中含有15个门类的细菌,优势菌群为变形菌门(50.94%),次优势菌群为拟杆菌门(46.16%); DJ13样品中含有20个门类的细菌,优势菌群为变形菌门(68.64%),次优势菌群为拟杆菌门(13.56%); DJ14样品中含有18个门类的细菌,优势菌群为蓝细菌门Cyanobacteria(41.07%),而变形菌门和拟杆菌门所占比例分别为36.96%和13.58%。由此表明,各样品中含有的菌群组成虽有差异,但变形菌门和拟杆菌门为优势菌,在样品中检测到的范围为50.54%—99.91%。
 |
| 图 3 样品中基于门水平的菌群相对丰度分析 Fig. 3 Relative abundance of phylum-level bacterial communities in different samples |
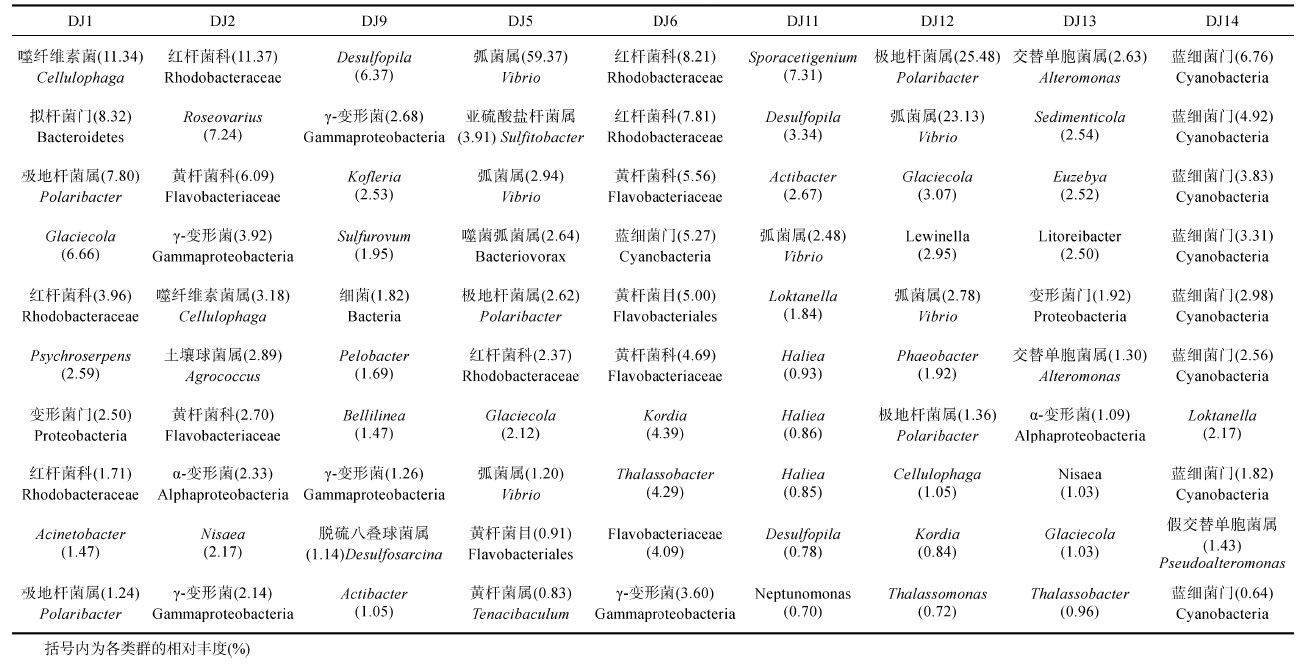
不同样品中所含菌群丰度最高的10个OUTs能够有助于了解样品中所含的主要细菌类型(表 3)。9个样品中所含菌群丰度最高的10个OTUs的序列经比对发现主要与变形菌门、拟杆菌门、放线菌门和蓝细菌门的细菌相似。其中,DJ1为噬纤维素菌属Cellulophaga(11.34%)、拟杆菌门Bacteroidetes(8.32%)、极地杆菌属Polaribacter(7.80%—1.24%)、Glaciecola(6.66%)、红杆菌科Rhodobacteraceae(3.96%)、Psychroserpens(2.59%—1.71%)、变形菌门Proteobacteria(2.50%)、不动杆菌属Acinetobacter(1.47%); DJ2为红杆菌科(11.37%)、Roseovarius(7.24%)、黄杆菌科Flavobacteriaceae(6.09%— 2.70%)、γ-变形菌Gammaproteobacteria(3.92%— 2.14%)、噬纤维素菌属(3.18%)、土壤球菌属Agrococcus(2.89%)、α-变形菌Aphlaproteobacteria(2.33%)、Nisaea(2.17%); DJ9为Desulfopila(6.37%)、γ-变形菌(2.68%—1.26%)、Kofleria(2.53%)、Sulfurovum(1.95%)、细菌Bacteria(1.82%)、Pelobacter(1.69%)、Bellilinea(1.47%)、脱硫八叠球菌属Desulfosarcina(1.14%)、Actibacter(1.05%); DJ5为弧菌属Vibrio(59.37%—1.20%)、亚硫酸盐杆菌属Sulfitobacter(3.91%)、噬菌弧菌属Bacteriovorax(2.64%)、极地杆菌属(2.62%)、红杆菌科(2.37%)、Glaciecola(2.12%)、黄杆菌目Flavobacteriales(0.91%)、黄杆菌属Tenacibaculum(0.83%); DJ6为红杆菌科(8.21%—7.81%)、黄杆菌科(5.56%—4.09%)、蓝细菌门Cyanobacteria(5.27%)、黄杆菌目(5.00%)、Kordia(4.39%)、Thalassobacter(4.29%)、γ-变形菌(3.60%); DJ11为Sporacetigenium(7.31%)、Desulfopila(3.34%—0.78%)、Actibacter(2.67%)、弧菌属(2.48%)、Loktanella(1.84%)、Haliea(0.93%—0.85%)、Neptunomonas(0.70%); DJ12有极地杆菌属(25.48%— 1.36%)、弧菌属(23.13%—2.78%)、Glaciecola(3.07%)、Lewinella(2.95%)、Phaeobacter(1.92%)、噬纤维素菌属(1.05%)、Kordia(0.84%)、Thalassomonas(0.72%); DJ13有交替单胞菌属Alteromonas(2.63%—1.30%)、Sedimenticola(2.54%)、Euzebya(2.52%)、Litoreibacter(2.50%)、变形菌(1.92%)、α-变形菌(1.09%)、Nisaea(1.03%)、Glaciecola(1.03%)、Thalassobacter(0.96%); DJ14为蓝细菌门(6.76%—0.64%)、Loktanella(2.17%)、假交替单胞菌属Pseudoalteromonas(1.43%)。值得注意的是,在DJ5和DJ12样品中弧菌属细菌含量较高,分别为59.37%—1.20%和23.13%—2.78%。
 |
下一代测序技术(Next Generation Sequence,NGS)主要是通过16S rRNA基因[细菌和古生菌的分子钟(Woese,1987)]分析环境中的群落组成,能够提供大量的微生物信息,因而被广泛应用于各种环境微生物的研究。有研究表明,长序列能够使分类比对更准确(Wang et al,2007)。罗氏454焦磷酸测序由于其能够得到相对较长的序列而被应用于许多微生物方面的研究(Herlemann et al,2011; Xia et al,2011)。Illumina测序平台虽然费用较454焦磷酸测序低且能够产生大量序列,但是由于得到的序列较短限制了其在微生物群落研究中的应用。然而,近年来Illumina测序平台逐渐提高了其测序性能,利用序列延伸法克服了所测序列较短的缺陷。目前,应用Illumina Miseq测序能够得到250bp或300bp左右的碱基长度的序列(Jeon et al,2015),使得该测序技术逐渐应用于微生物群落的研究(Bell et al,2013; Yang et al,2015)。本研究利用基于Illumina Miseq平台的高通量测序分析了大连地区3个养殖公司的刺参养殖环境菌群多样性,9个样品所得有效序列为26503—37825条,可归为1502—5741个分类操作单元(OTUs),进一步分析表明样品9个样品中除检测以往报道过的刺参养殖环境中的细菌变形菌门、拟杆菌门、绿弯菌门、浮霉菌门、蓝细菌门、酸杆菌门、疣微菌门、厚壁菌门和放线菌门(Li et al,2010; 王轶南等,2010; 关晓燕等,2010; 李建光等,2014)外,还检测到嗜热丝菌门、古细菌、柔膜菌门、纤维杆菌门、互养菌门、硝化螺旋菌门、迷踪菌门、装甲菌门、衣原体门、异常球菌-栖热菌门、绿菌门、芽单胞菌门、梭杆菌门、黏胶球形菌门、产水菌门、脱铁杆菌门和螺旋体门等17个鲜见报道的类群,其在9个样品中的检测量均不足1%,表明基于Illumina Miseq平台的高通量测序可作为全面了解环境中菌群多样性的一种有效研究手段。
多样性指数和稀缺性曲线进一步分析,3个公司的刺参养殖环境沉积物中群落多样性较其刺参养殖水环境中菌群多样性高。与Lozupone等(2007)的研究结果即沉积物中微生物多样性较其它环境类型中微生物多样性较高相一致。本研究结果显示变形菌门和拟杆菌门为各样品的优势菌。变形菌门类的细菌在刺参养殖环境中普遍存在(Li et al,2010b; 任利华等,2015)。本研究各样品中的变形菌主要为γ-变形菌和α-变形菌。γ-变形菌中主要包括Glaciecola属、不动杆菌属、弧菌属、单胞菌属等。γ-变形菌是海洋中普遍存在的细菌,多样性很高,数量也很大,尤其在营养物质含量高的水域,都是主要的优势细菌,大多数都能够培养(Ivanova et al,2003; Payne et al,2006)。通常在厌氧环境中,部分γ-变形菌可与该环境中的动物形成共生关系,对环境中的碳、硫循环起着重要作用(Bakunina et al,1999; Huber et al,2004)。α-变形菌主要属于红细菌目、红细菌科的细菌,这类细菌能够通过光合作用进行生长代谢,CO2和氮的固定(白洁等,2009),因此在刺参养殖环境系统碳、氮循环中发挥着重要作用。拟杆菌门的细菌与DNA、脂类和蛋白质等有机物质的转换密切相关,这些有机物质的吸收和利用是各种水体环境中碳循环的重要组成部分(Cottrell et al,2000; O'Sullivan et al,2002)。在养殖过程中,对刺参进行喂食的有机饲料,未被完全消化的饲料、粪便在水体中未能及时被分解,便会沉积下来(吴庆龙等,1995),会造成池底拟杆菌细菌含量增加(Rosselló-Mora et al,1999)。3个公司刺参养殖环境水体和沉积物中都发现了该类群的细菌,这说明这些养殖环境中存在着较为活跃的物质循环。此外,蓝细菌是海洋生态系统和初级生产力的重要组成部分,能通过同化CO2来生成O2,在好氧条件下固氮,在黑暗中通过厌氧呼吸来还原硫(白洁等,2009),在水样和沉积物样品中均检测到蓝细菌的存在,尤其是旅顺养殖公司的刺参底播养殖沉积物(DJ14)中蓝细菌占41.07%。
近年来,关于弧菌引起的刺参病害的研究时常报道。张春云等(2006)和杨求华等(2014)从患有“腐皮综合症”的刺参病灶处分离的致病菌分别为灿烂弧菌V. splendidus和塔式弧菌V. tubiashii,腐皮综合症发病急,病程短,死亡率高是刺参幼参和成参时期危害最为严重的疾病(张鹏等,2013)。王印庚等(2007)在仿刺参苗种培育期间从患“烂胃病”的耳状幼体中分离到1株细菌,鉴定为灿烂弧菌并视为耳状幼体“烂胃病”的致病原之一。杨嘉龙等(2007)从患“溃疡病”的仿刺参病灶处分离的致病菌被鉴定为溶藻弧菌V. alginolyticus。马悦欣等(2006)从患急性口围肿胀症的刺参的口围、体表和呼吸树中分离得到的优势菌经人工感染试验证实,五株引起溃烂病的病原菌中有四株属于弧菌属。Deng等(2009)从患有“腐皮综合病”的刺参体表和排脏中分离纯化得到8株优势菌,经研究为致病菌,进一步鉴定分别为弧菌属、发光杆菌属Photobacterium、节杆菌属Arthrobacter、葡萄球菌属Staphylococcus的细菌,其中有4株为弧菌。弧菌属虽是正常菌属的一部分,但是在某些特殊条件发生,如刺参受到应激影响或体表损伤时,会迅速感染刺参(Thompson et al,2004; Thompson et al,2005)。本研究中,庄河和旅顺养殖公司的室内养殖池塘中检测有大量的弧菌属的细菌,虽然两养殖公司的刺参未有疾病的暴发,但是为了养殖刺参的健康生长,应注意刺参养殖加强管理,观察刺参活动状态,摄食与粪便情况,保持池底清洁,定时测量水质指标。 4 结论
刺参养殖作为我国水产养殖的重要产业和北方地区养殖结构调整的优良水产品种,具有较高的经济效益和发展潜力。近年来海参疾病的发生波及范围广,死亡率高且有连年加重之势,病害的泛滥成为制约其发展的主要限制因素之一,因此解决当前病害问题成为一种迫切的市场需求。
目前关于刺参养殖环境中菌群结构的研究已有报道,前文已作阐述。病害的发展是一个复杂的过程,微生物群落在这一过程中数量和结构也在不断变化,与刺参的健康状况紧密相关。本研究采用高通量测序技术分析了大连地区3个养殖公司刺参养殖环境中室内养殖池塘水体、室外养殖水体和沉积物中菌群结构特征,揭示了刺参池塘养殖环境中的菌群多样性和丰度,比较了其菌群结构及差异,为刺参健康养殖提供参考。
| 马悦欣,徐高蓉,张恩鹏等,2006.仿刺参幼参急性口围肿胀症的细菌性病原.水产学报, 30(3):377-382 |
| 王印庚,孙素凤,荣小军,2007.仿刺参幼体烂胃病及其致病原鉴定.中国水产科学, 13(6):908-916 |
| 王轶南,朱世伟,常亚青,2010.刺参肠道及养殖池塘菌群组成的PCR-DGGE指纹图谱分析.渔业科学进展, 31(3):119-122 |
| 白洁,李海艳,赵阳国,2009.黄海北部不同站位海洋细菌群落分布特征.微生物学报, 49(3):343-350 |
| 任利华,李斌,孙国华等,2015. 16S rDNA克隆文库解析仿刺参(Apostichopus japonicus)苗种培育池中生物絮团的细菌群落结构.海洋与湖沼, 46(1):197-205 |
| 关晓燕,周遵春,陈仲等,2010.应用PCR-DGGE指纹技术分析高温季节仿刺参养殖水环境中菌群多样性.海洋湖沼通报, (1):82-88 |
| 李建光,徐永平,李晓宇等,2014.不同养殖季节仿刺参肠道与养殖环境中菌群结构的特点.水产科学, 33(9):562-568 |
| 杨求华,葛辉,方旅平等,2014.池塘养殖刺参病原菌塔式弧菌的分离与鉴定.南方水产科学, 10(4):45-51 |
| 杨嘉龙,周丽,绳秀珍等,2007.养殖刺参溃疡病病原菌RH2的鉴定及其生物学特性分析.水产学报, 31(4):504-511 |
| 吴庆龙,陈开宁,高光等,1995.大水面网围精养对水环境的影响及其对策.水产学报, 19(4):343-349 |
| 张鹏,李成华,李晔等,2013.刺参(Apostichopus japonicus)腐皮综合症发生相关蛋白的分离与鉴定.海洋与湖沼, 44(3):741-746 |
| 张春云,王印庚,荣小军等,2006.养殖刺参腐皮综合征病原菌的分离与鉴定.水产学报, 30(1):118-123 |
| Amann R I, Ludwig W, Schleifer K H,1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiological Reviews, 59(1):143-169 |
| Auguet J-C, Barberan A, Casamayor E O,2009. Global ecological patterns in uncultured Archaea. The International Society for Microbial Ecology Journal, 4(2):182-190 |
| Bakunina I, Shevchenko L S, Nedashkovskaia O I et al,1999. Screening of marine bacteria for fucoidan hydrolases. Mikrobiologiia, 69(3):370-376 |
| Bell T H, Yergeau E, Maynard C et al,2013. Predictable bacterial composition and hydrocarbon degradation in Arctic soils following diesel and nutrient disturbance. The International Society for Microbial Ecology Journal, 7(6):1200-1210 |
| Bordbar S, Anwar F, Saari N,2011. High-value components and bioactives from sea cucumbers for functional foods——A review. Marine Drugs, 9(12):1761-1805 |
| Caporaso J G, Lauber C L, Walters W A et al,2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America, 108(S1):4516-4522 |
| Cottrell M T, Kirchman D L,2000. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low-and high-molecular-weight dissolved organic matter. Applied and Environmental Microbiology, 66(4):1692-1697 |
| Deng H, He C B, Zhou Z C et al,2009. Isolation and pathogenicity of pathogens from skin ulceration disease and viscera ejection syndrome of the sea cucumber Apostichopus japonicus. Aquaculture, 287(1-2):18-27 |
| Edgar R C, Haas B J, Clemente J C et al,2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16):2194-2200 |
| Herlemann D P, Labrenz M, Jürgens K et al,2011. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. The International Society for Microbial Ecology Journal, 5(10):1571-1579 |
| Huber I, Spanggaard B, Appel K F et al,2004. Phylogenetic analysis and in situ identification of the intestinal microbial community of rainbow trout (Oncorhynchus mykiss, Walbaum). Journal of Applied Microbiology, 96(1):117-132 |
| Hugenholtz P, Hooper S D, Kyrpides N C,2009. Focus:Synergistetes. Environmental Microbiology, 11(6):1327-1329 |
| Ivanova E P, Sawabe T, Zhukova N V et al,2003. Occurrence and diversity of mesophilic Shewanella strains isolated from the North-West Pacific Ocean. Systematic and Applied Microbiology, 26(2):293-301 |
| Jeon Y-S, Park S-C, Lim J et al,2015. Improved pipeline for reducing erroneous identification by 16S rRNA sequences using the Illumina Miseq platform. Journal of Microbiology, 53(1):60-69 |
| Li H, Qiao G, Li Q et al,2010a. Biological characteristics and pathogenicity of a highly pathogenic Shewanella marisflavi infecting sea cucumber, Apostichopus japonicus. Journal of Fish Diseases, 33(11):865-877 |
| Li Q F, Zhang Y, Juck D et al,2010b. Phylogenetic analysis of bacterial communities in the shrimp and sea cucumber aquaculture environment in northern China by culturing and PCR-DGGE. Aquaculture International, 18(6):977-990 |
| Lozupone C A, Knight R,2007. Global patterns in bacterial diversity. Proceedings of the National Academy of Sciences of the United States of America, 104(27):11436-11440 |
| Luo P, Hu C Q, Xie Z Y et al,2006. PCR-DGGE analysis of bacterial community composition in brackish water Litopenaeus vannamei culture system. Journal of Tropical Oceanography, 25(2):49-53 |
| O'Sullivan L A, Weightman A J, Fry J C,2002. New degenerate Cytophaga-Flexibacter-Bacteroides-specific 16S ribosomal DNA-targeted oligonucleotide probes reveal high bacterial diversity in River Taff epilithon. Applied and Environmental Microbiology, 68(1):201-210 |
| Paerl H W, Dyble J, Moisander P H et al,2003. Microbial indicators of aquatic ecosystem change:current applications to eutrophication studies. FEMS Microbiology Ecology, 46(3):233-246 |
| Payne M S, Hall M R, Bannister R et al,2006. Microbial diversity within the water column of a larval rearing system for the ornate rock lobster (Panulirus ornatus). Aquaculture, 258(1-4):80-90 |
| Purcell S W, Hair C A, Mills D J,2012. Sea cucumber culture, farming and sea ranching in the tropics:progress, problems and opportunities. Aquaculture, 368-369:68-81 |
| Ren G D, Ren W J, Teng Y T et al,2015. Evident bacterial community changes but only slight degradation when polluted with pyrene in a red soil. Frontiers in Microbiology, 6:22 |
| Rosselló-Mora R, Aman R,2001. The species concept for prokaryotes. FEMS Microbiology Reviews, 25(1):39-67 |
| Rosselló-Mora R, Thamdrup B, Schäfer H et al,1999. The response of the microbial community of marine sediments to organic carbon input under anaerobic conditions. Systematic and Applied Microbiology, 22(2):237-248 |
| Schloss P D, Westcott S L, Ryabin T et al,2009. Introducing mothur:open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23):7537-7541 |
| Thompson F L, Iida T, Swings J,2004. Biodiversity of vibrios. Microbiology and Molecular Biology Reviews, 68(3):403-431 |
| Thompson J R, Pacocha S, Pharino C et al,2005. Genotypic diversity within a natural coastal bacterioplankton population. Science, 307(5713):1311-1313 |
| Wang Q, Garrity G M, Tiedje J M et al,2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73(16):5261-5267 |
| Wang Y G, Zhang C Y, Rong X J et al,2005. Diseases of cultured sea cucumber, Apostichopus japonicus, in China. FAO Fisheries Technical Paper, 297-310 |
| Woese C R,1987. Bacterial evolution. Microbiological Reviews, 51(2):221-271 |
| Xia W W, Zhang C X, Zeng X W et al,2011. Autotrophic growth of nitrifying community in an agricultural soil. The International Society for Microbial Ecology Journal, 5(7):1226-1236 |
| Yang Y Y, Wang Z, He T et al,2015. Sediment Bacterial Communities Associated with Anaerobic Biodegradation of Bisphenol A. Microbial Ecology, 70(1):97-104 |
| Youssef N, Sheik C S, Krumholz L R et al,2009. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Applied and Environmental Microbiology, 75(16):5227-5236 |