 2023, Vol. 54
2023, Vol. 54中国海洋湖沼学会主办。
文章信息
- 许涛, 孔令锋. 2023.
- XU Tao, KONG Ling-Feng. 2023.
- 软体动物线粒体基因组不同组装策略的比较研究
- COMPARATIVE ANALYSIS OF DIFFERENT STRATEGIES OF MOLLUSCAN MITOCHONDRIAL GENOME ASSEMBLY
- 海洋与湖沼, 54(2): 537-549
- Oceanologia et Limnologia Sinica, 54(2): 537-549.
- http://dx.doi.org/10.11693/hyhz20220700190
文章历史
-
收稿日期:2022-07-20
收修改稿日期:2022-10-09




2. 中国海洋大学三亚海洋研究院 海南三亚 572000;
3. 崂山实验室海洋渔业科学与食物产出过程功能实验室 山东青岛 266237
2. Sanya Oceanographic Institution, Ocean University of China, Sanya 572000, China;
3. Laboratory for Marine Fisheries Science and Food Production Processes, Laoshan Laboratory, Qingdao 266237, China
线粒体基因组序列长度较短, 组成和结构相对稳定, 非编码区较少, 极少发生重组, 进化速度快, 不仅包含序列信息, 还有着丰富的基因结构信息(Doiron et al, 2002; Gissi et al, 2008; Wei et al, 2010; Li et al, 2012), 现已成为研究群体遗传分化(Arndt et al, 1998; 邢晶晶, 2002; Wang et al, 2015)、基因进化(Perseke et al, 2010)、物种起源(Saccone et al, 1999)、疾病诊断(Hart et al, 2013)、法医学鉴定(Davis et al, 2015)、分子标记挖掘(Galaska et al, 2019)、系统发育(Miya et al, 2001; Osigus et al, 2013; Li et al, 2015; Mikkelsen et al, 2018)等领域的重要工具。
早期线粒体基因组的获得主要是对物理分离出的线粒体DNA的克隆文库进行Sanger测序, 即将细胞的不同组分通过离心(氯化铯密度梯度离心或差速离心法)或碱裂解法分开, 获得高纯度的线粒体DNA, 然后用限制性核酸内切酶进行部分或完全酶切, 或者用不同强度的超声波随机打断成短片段, 最后将这些短片段克隆到质粒载体(Plasmid Vectors)中进行测序(Tamura et al, 1988; 沙淼等, 2013)。该方法充分保证了线粒体序列的准确性(Tzeng et al, 1992; 鲁成等, 2002), 但冗繁的实验步骤, 对样品的高需求量以及质量的高要求等限制了该方法的大规模应用(沙淼等, 2013; 李天杰等, 2016)。1985年, 聚合酶链式反应PCR (Polymerase Chain Reaction)技术首次被提出(Saiki et al, 1985), Sanger测序PCR扩增产物的方法自此成为了获取线粒体基因组的主流, 该方法是指通过设计特异性引物经Long PCR (Cheng et al, 1994)扩增出线粒体基因组的大片段, 结合引物步移法(primer walking)进行Sanger测序, 最后对测序得到的不同片段进行拼接得到完整的线粒体基因组序列。该方法获得的线粒体基因组准确性高, 对DNA母液的需求量少, 规避了线粒体假基因的干扰以及物理纯化线粒体DNA的操作步骤(沙淼等, 2013), 即使是测序技术快速发展的今天该方法仍然占据了很大的市场。但该方法在大规模应用时逐渐显露出不足之处, 如通量低、花费高、耗时长、特异性引物设计难度较大以及对DNA质量的严格要求限制了保存不当的样品的线粒体基因组的测定等(Hofreiter et al, 2001; Orlando et al, 2015)。
2012年, 基因组浅层测序技术被首次提出(Straub et al, 2012), 指通过鸟枪测序法(shotgun sequencing)获得全基因组较低测序深度(low-passing)数据, 能够恢复细胞器基因组以及低拷贝核基因的序列片段(Sarmashghi et al, 2019)。该技术通量高、成本低、速度快, 且无需设计引物, 在过去的十年间得到了广泛应用。如何在避免序列长、拷贝数高的核内线粒体假基因(nuclear mitochondrial pseudogenes, 也称Numts)污染的情况下从测序数据中准确、快速的抓取线粒体reads, 并成功组装出完整的线粒体基因组序列成为了亟需解决的关键问题(李艳等, 2012; Li et al, 2012)。目前从基因组浅层测序数据中获取线粒体基因组的主流的组装策略主要分为有参考序列组装策略以及从头(de novo)组装策略(Hunter et al, 2015; Machado et al, 2016)。有参考序列组装策略是指将测序获得的短读长(short reads)映射到参考序列上, 根据近缘物种线粒体基因组序列的相似性捕获线粒体reads并不断延伸, 直至获得完整的线粒体基因组(Hunter et al, 2015), 该方法速度快, 但序列准确性完全依赖于参考序列(The 1000 Genomes Project Consortium et al, 2010)。其中, 线粒体基因组作为参考序列时, 序列的延伸是通过映射到参考序列上的线粒体reads间的相互重叠完成, 而在以线粒体基因组序列片段为参考序列的组装中则是通过将映射捕获的高覆盖度且连续的线粒体reads拼接成重叠群, 成为新一轮映射的参考序列, 以此反复将原始测序数据映射到新的参考序列上, 直至获得完整的线粒体基因组(匡卫民等, 2019)。从头组装策略与有参组装策略有很大的区别, 从头组装策略是将所有短读长拼接成多条重叠群序列(Hunter et al, 2015), 根据线粒体基因组测序深度高的特点确定可能的线粒体重叠群, 然后将测序数据反复映射到这些重叠群, 并不断延长(匡卫民等, 2019), 该方法组装时运算量大(Li et al, 2012)。目前可公开获得并且免费使用的主流的线粒体基因组组装软件有: Ray (Boisvert et al, 2010)、SPAdes (Bankevich et al, 2012)、MitoZ (Meng et al, 2019)、NOVOPlasty (Dierckxsens et al, 2017)、GetOrganelle (Jin et al, 2020), 以及最新发表的基于从头组装策略的线粒体基因组组装软件MEANGS (Song et al, 2022)。其中NOVOPlasty基于参考序列的组装策略, 其余5个软件均基于从头组装策略, 但是开发者对组装流程进行了不同方向的优化。在之前的研究中, Ray、SPAdes、MitoZ、NOVOPlasty、GetOrganelle成功用于组装不同动物类群的线粒体基因组, 并基于线粒体基因组序列进一步开展了相关研究(Li et al, 2015; Kong et al, 2020; Wang et al, 2021; Zhao et al, 2021; Kitano et al, 2022; Zhang et al, 2022)。
软体动物是动物界第二大门类, 从热带到极地地区均有分布, 栖息于深海热液口、潮间带、陆地等各种各样的生境, 是重要的食物和装饰品来源, 经济价值高, 富含药用、营养价值(Ponder et al, 2020), 广泛应用于细胞生物学、神经生物学、生理学、行为学、进化论、群体遗传学和材料科学研究(Liu et al, 2021)。软体动物的线粒体基因组在大小、结构和功能方面存在巨大变异(Ghiselli et al, 2021), 但是目前仍然缺乏专门为软体动物设计的线粒体基因组组装软件。本研究中利用目前主流的组装软件Ray、SPAdes、MitoZ、GetOrganelle、MEANGS和NOVOPlasty, 通过比较各个软件的运行时间、基因组覆盖度、线粒体重叠群、结果文件的大小以及线粒体基因组全序列的质量, 测试它们在软体动物主要类群腹足纲Gastropoda、双壳纲Bivalvia、头足纲Cephalopoda、多板纲Polyplacophora中的组装效果, 以期优化软体动物不同类群的线粒体基因组的获取流程, 为后续开展线粒体基因组的相关研究奠定基础。
1 材料与方法 1.1 实验数据本研究使用的实验数据信息见表 1, 通过分析NCBI中已公开发表的软体动物主要类群的线粒体基因组数据, 从隶属于软体动物的多板纲、双壳纲、腹足纲以及头足纲中各选择了3个代表物种。其中由于非编码区(non-coding regions)扩增和转座(transposition)等原因(Smith et al, 2007; Sun et al, 2016), 双壳贝类一些种类的线粒体DNA全长是其他两侧对称动物的2~3倍, 如魁蚶Scapharca broughtonii (46 985 bp; Liu et al, 2013)、夹粗饰蚶Anadara vellicata (34 147 bp; Sun et al, 2015)、麦哲伦扇贝Placopecten magellanicus (30 680~40 725 bp; Smith et al, 2007)、Bryopa lata (> 31 969 bp; Williams et al, 2017), 因此在双壳纲中我们选择的3个物种分别是经测定有着常见线粒体基因组大小(17 903 bp)的虾夷蚶蜊Glycymeris yessoensis (Kong et al, 2020), 经测定线粒体基因组大小为38 672 bp的结蚶Tegillarca nodifera以及线粒体基因组大小高达56 170 bp的毛蚶Scapharca kagoshimensis (Kong et al, 2020)。其余软体动物中除Lottia digitalis (26 835 bp)和帝巨奥氏蛞蝓Megaustenia imperator (34 791 bp)外线粒体基因组大小均在13 000~21 000 bp, 本研究中多板纲(14 936~16 573 bp)、腹足纲(13 453~20 092 bp)以及头足纲(14 654~18 999 bp)的代表物种的选择涉及线粒体基因组各个范围(如腹足纲中选择的斗嫁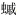
| 纲 | 物种名 | 拉丁名 | 采集地点 | 参考物种名 | 参考物种拉丁名 | 测序平台 |
| 双壳纲 | 虾夷蚶蜊 | Glycymeris yessoensis | 江苏连云港 | — | Lunarca ovails | Illumina HiSeq X |
| 毛蚶 | Scapharca kagoshimensis | 广西北海 | 胀毛蚶 | Scapharca globosa | Illumina HiSeq X | |
| 结蚶 | Tegillarca nodifera | 广西防城港 | — | Tegillarca sp. | Illumina HiSeq X | |
| 多板纲 | 史氏宽板石鳖 | Placiphorella stimpsoni | 福建漳州 | — | Katharina tunicata | Illumina HiSeq X |
| 红条毛肤石鳖 | Acanthochitona rubrolineata | 福建漳州 | — | Acanthochitona avicula | Illumina HiSeq X | |
| 琉球花棘石鳖 | Acanthopleura loochooana | 福建漳州 | — | Acanthopleura echinata | Illumina HiSeq X | |
| 腹足纲 | 斗嫁 |
Cellana grata | 福建宁德 | — | Nacella magellanica | Illumina NovaSeq 6000 |
| 三列扭柱螺 | Tectus triserialis | 海南三沙 | 塔形扭柱螺 | Tectus pyramis | Illumina HiSeq X | |
| — | Pseudosuccinea columella | 静水椎实螺 | Lymnaea stagnalis | |||
| 头足纲 | 菱鳍鱿 | Thysanoteuthis rhombus | 中国南海 | 阿根廷滑柔鱼 | Illex argentinus | Illumina NovaSeq 6000 |
| 鹦鹉螺 | Nautilus pompilius | 大脐鹦鹉螺 | Nautilus macromphalus | |||
| 图氏后乌贼 | Metasepia tullbergi | 白斑乌贼 | Sepia latimanus |
分别应用软件NOVOPlasty (Dierckxsens et al, 2017)、GetOrganelle (Jin et al, 2020)、MitoZ (Meng et al, 2019)、Ray (Boisvert et al, 2010)、SPAdes (Bankevich et al, 2012)和MEANGS (Song et al, 2022)组装软体动物主要类群代表物种的线粒体基因组。组装软件的运行基于实验室现有服务器, 服务器运行在64 bit模式下, CPU型号为Intel(R) Xeon(R) Gold 6132 CPU @ 2.60 GHz, Linux内核信息Linux localhost.localdomain 3.10.0-1062.9.1.el7.x86_64 #1 SMP Fri Dec 6 15:49:49 UTC 2019 x86_64 x86_64 x86_64 GNU/Linux, 物理内存共503 G, 硬盘交换分区共127 G。组装时除NOVOPlasty默认用1个线程(thread)运行外, 其余软件均使用8个线程运行。Ray的k-mer设定为31, SPAdes的k-mer设定为21、33、55、77, GetOrganelle在调用SPAdes进行从头组装时的k-mer设定为21、45、65、85、105, 最大组装延伸轮数为15, 以上均为软件默认或推荐参数。其中基于参考序列组装策略的NOVOPlasty所用参考序列在表 1中已注明。Ray和SPAdes组装后并不能直接得到线粒体基因组序列, 还需要将所得骨架(Scaffolds)建库, 借助参考序列(见表 1)进一步BLAST获得线粒体基因组序列。组装得到的线粒体基因组均通过MITOS (Bernt et al, 2013)进行注释以检查是否存在基因缺失。
1.3 组装质量的评估关于组装质量的评估比较, 我们评价的指标基本参照了Dierckxsens等(2017), 具体包括软件运行时长、重叠群数目、基因组覆盖度、基因组准确性、结果文件的存储空间占用。本研究中组装时间的确定借助组装软件在Linux系统中运行时用命令time获得; 重叠群的数目在线粒体基因组最后的组装结果中可以直接明确; 结果文件的大小直接在Linux环境下用“ls -al”查看; 缺失基因的确定通过MITOS初步注释, 并经Open Reading Frame Finder (https://www.ncbi.nlm.nih.gov/orffinder/)、ARWEN (Laslett et al, 2008), 以及已发表的线粒体基因组分别进一步检查确定。对线粒体基因组序列质量的判断主要是借助软件Quast (Gurevich et al, 2013), Quast用于评价线粒体基因组序列质量时分为有参考序列和无参考序列两种方法。QUAST在无参的情况下缺乏可供评判不同线粒体基因组序列质量的参数, 为了更好评价各个软件的组装效果, 本研究使用有参考线粒体基因组的方法, 参考序列首选相应物种在NCBI中已公开完整的线粒体基因组序列, 并经MitoZ中的circle_check.py脚本确定是否为环状, 并去除两端的重复序列作为参考序列。当两端重复序列太短或者本身并非完整的线粒体基因组导致circle_check.py脚本认为该序列并非环状时, 使用软件MEGA 5.0 (Tamura et al, 2011)比较NCBI中已发表的线粒体基因组序列以及各个软件组装出来的结果, 确定最终的参考序列。通过QUAST结果报告中Genome fraction确定基因组覆盖度, 即组装好的序列占参考序列的百分比, 通过报告中的Genome coverage确定准确性, 即组装好的序列去除错配的位点后占参考基因组的百分比。由于NOVOPlasty的组装结果中仅包含组装的结果文件和日志, 存储空间占用十分小, 因此, 当NOVOPlasty的结果文件存储空间占用小于1 MB时, 为了结果文件存储空间的单位的统一, 在图表中均记为0.01 GB。
为更加直观地比较各个软件的组装效果, 组装结果以表格和柱状图两种形式呈现。其中在柱状图的绘制中, 基因组覆盖度、准确性、运行时间、结果文件存储空间占用、线粒体重叠群数目的最高得分分别设置在100%、100%、0 min、0 GB、1条, 所有百分比都保留至小数点后两位。
2 结果在NOVOPlasty组装软体动物主要类群代表物种的组装结果中(表 2, 图 1), 除在头足纲鹦鹉螺中的组装时间为62 min外, 其余为6~18 min, 结果文件存储空间占用均小于0.01 GB。在组装质量方面, NOVOPlasty在组装双壳纲的毛蚶时报错, 无法完成组装, 在组装头足纲的菱鳍鱿时, 基因组覆盖度为86.53%, 基因组准确度仅为77.09%, 在组装头足类的图氏后乌贼的结果中, 基因组覆盖度仅为61.41%, 但是准确度达100%, 在本研究中的其他软体动物类群中, NOVOPlasty组装获得线粒体基因序列基因组覆盖度在99.11%~100%, 基因组准确度为99.32%~100%。
| 纲 | 物种名 | 运行时长/min | 重叠群数目/条 | 基因组覆盖度/% | 基因组准确度/% | 结果文件存储空间占用/GB |
| 双壳纲 | 虾夷蚶蜊 | 11 | 1 | 100 | 100 | 0.01 |
| 结蚶 | 14 | 3 | 99.11 | 99.87 | 0.01 | |
| 毛蚶 | 报错并自动退出组装 | |||||
| 多板纲 | 史氏宽板石鳖 | 12 | 1 | 100 | 100 | 0.01 |
| 红条毛肤石鳖 | 13 | 1 | 100 | 100 | 0.01 | |
| 琉球花棘石鳖 | 11 | 1 | 99.64 | 100 | 0.01 | |
| 腹足纲 | 斗嫁 |
10 | 1 | 100 | 100 | 0.01 |
| 三列扭柱螺 | 16 | 2 | 100 | 100 | 0.01 | |
| Pseudosuccinea columella | 18 | 1 | 100 | 99.32 | 0.01 | |
| 头足纲 | 菱鳍鱿 | 13 | 5 | 86.53 | 77.09 | 0.01 |
| 鹦鹉螺 | 62 | 1 | 100 | 100 | 0.01 | |
| 图氏后乌贼 | 6 | 1 | 61.41 | 100 | 0.01 | |
 |
| 图 1 NOVOPlasty的基准研究得分图 Fig. 1 Score graph based on the benchmark study of NOVOPlasty 注: 每个组装软件的得分都是基于组装结果, 并以百分比表示。百分比越高, 组装结果越好 |
在MitoZ组装软体动物主要类群代表物种的组装结果中(表 3, 图 2), MitoZ的运行时长为57 min (图氏后乌贼)~396 min (菱鳍鱿)。结果文件占用1.38 GB (图氏后乌贼)~9.50 GB (菱鳍鱿)。在组装质量方面, MitoZ在组装双壳纲的毛蚶和头足纲的鹦鹉螺时发生报错, 未能完成组装, 双壳纲虾夷蚶蜊的组装结果中, 尽管基因组覆盖度为100%, 但是基因组准确度为0%, 腹足纲P. columella的组装结果中基因组覆盖度和准确度均为0%, 在双壳类的结蚶和头足纲的菱鳍鱿的组装结果中基因组覆盖度分别为83.88%和40.72%, 但准确度分别为99.33%和100%。在本研究中组装的其他软体动物类群中基因组覆盖度为97.53%~100%, 准确度均为100%。
| 纲 | 物种名 | 运行时长/min | 重叠群数目/条 | 基因组覆盖度/% | 基因组准确度/% | 结果文件存储空间占用/GB |
| 双壳纲 | 虾夷蚶蜊 | 243 | 1 | 100 | 0 | 4.90 |
| 结蚶 | 137 | 1 | 83.88 | 99.33 | 6.50 | |
| 毛蚶 | 报错并自动退出组装 | |||||
| 多板纲 | 史氏宽板石鳖 | 160 | 1 | 100 | 100 | 3.78 |
| 红条毛肤石鳖 | 167 | 1 | 100 | 100 | 4.08 | |
| 琉球花棘石鳖 | 126 | 1 | 99.64 | 100 | 8.40 | |
| 腹足纲 | 斗嫁 |
126 | 1 | 100 | 100 | 3.29 |
| 三列扭柱螺 | 203 | 1 | 99.05 | 100 | 4.95 | |
| Pseudosuccinea columella | 225 | 1 | 0 | 0 | 6.56 | |
| 头足纲 | 菱鳍鱿 | 396 | 1 | 40.72 | 100 | 9.50 |
| 鹦鹉螺 | 报错并自动退出组装 | |||||
| 图氏后乌贼 | 57 | 1 | 97.53 | 100 | 1.38 | |
 |
| 图 2 MitoZ的基准研究得分图 Fig. 2 Score graph based on the benchmark study of MitoZ 注: 每个组装软件的得分都是基于组装结果, 并以百分比表示。百分比越高, 组装结果越好 |
在GetOrganelle组装软体动物主要类群代表物种的组装结果中(表 4, 图 3), GetOrganelle的运行时长为57 min (斗嫁
| 纲 | 物种名 | 运行时长/min | 重叠群数目/条 | 基因组覆盖度/% | 基因组准确度/% | 结果文件存储空间占用/GB |
| 双壳纲 | 虾夷蚶蜊 | 71 | 1 | 100 | 100 | 0.03 |
| 结蚶 | 报错并自动退出组装 | |||||
| 毛蚶 | 报错并自动退出组装 | |||||
| 多板纲 | 史氏宽板石鳖 | 159 | 1 | 100 | 100 | 4.08 |
| 红条毛肤石鳖 | 208 | 1 | 100 | 100 | 6.37 | |
| 琉球花棘石鳖 | 286 | 5 | 70.79 | 100 | 6.77 | |
| 腹足纲 | 斗嫁 |
57 | 1 | 100 | 100 | 0.74 |
| 三列扭柱螺 | 143 | 1 | 98.38 | 100 | 1.26 | |
| Pseudosuccinea columella | 241 | 2 | 100 | 99.52 | 5.17 | |
| 头足纲 | 菱鳍鱿 | 81 | 1 | 100 | 100 | 0.02 |
| 鹦鹉螺 | 报错并自动退出组装 | |||||
| 图氏后乌贼 | 189 | 1 | 100 | 100 | 2.46 | |
 |
| 图 3 GetOrganelle的基准研究得分图 Fig. 3 Score graph based on the benchmark study of GetOrganelle 注: 每个组装软件的得分都是基于组装结果, 并以百分比表示。百分比越高, 组装结果越好 |
在MEANGS组装软体动物主要类群代表物种的组装结果中(表 5, 图 4), MEANGS的运行时长为54 min (图氏后乌贼)~252 min (鹦鹉螺)。结果文件存储空间占用3.09 GB (图氏后乌贼)~14.9 GB (鹦鹉螺)。在组装质量方面, MEANGS组装双壳纲的毛蚶的结果中, 基因组覆盖度为43.36%, 基因组准确度仅为37.52%, 在双壳纲结蚶、多板纲琉球花棘石鳖和头足纲菱鳍鱿的组装结果中, 基因组覆盖度分别为34.51%、78.90%、75.63%, 但是基因组准确度为99.95%~100%。在其他双壳类、石鳖、腹足类以及头足类的组装结果中, 基因组覆盖度为98.49%~100%, 基因组准确度为99.11%~100%。
| 纲 | 物种名 | 运行时长/min | 重叠群数目/条 | 基因组覆盖度/% | 基因组准确度/% | 结果文件存储空间占用/GB |
| 双壳纲 | 虾夷蚶蜊 | 117 | 1 | 100 | 100 | 9.69 |
| 结蚶 | 100 | 8 | 34.51 | 99.95 | 7.84 | |
| 毛蚶 | 115 | 3 | 43.46 | 37.52 | 7.63 | |
| 多板纲 | 史氏宽板石鳖 | 111 | 1 | 99.97 | 99.95 | 8.36 |
| 红条毛肤石鳖 | 125 | 1 | 100 | 100 | 8.45 | |
| 琉球花棘石鳖 | 89 | 2 | 78.90 | 100 | 7.96 | |
| 腹足纲 | 斗嫁 |
99 | 1 | 100 | 100 | 7.34 |
| 三列扭柱螺 | 133 | 1 | 99.95 | 100 | 10.10 | |
| Pseudosuccinea columella | 187 | 1 | 100 | 99.54 | 12.50 | |
| 头足纲 | 菱鳍鱿 | 130 | 3 | 75.63 | 100 | 10.00 |
| 鹦鹉螺 | 252 | 1 | 100 | 99.20 | 14.90 | |
| 图氏后乌贼 | 54 | 1 | 98.49 | 99.11 | 3.09 |
 |
| 图 4 MEANGS的基准研究得分图 Fig. 4 Score graph based on the benchmark study of MEANGS 注: 每个组装软件的得分都是基于组装结果, 并以百分比表示。百分比越高, 组装结果越好 |
在Ray组装软体动物主要类群代表物种的组装结果中(表 6, 图 5), Ray的运行时长为254 min (图氏后乌贼)~2659 min (鹦鹉螺)。结果文件存储空间占用3.43 GB (图氏后乌贼)~18.4 GB (菱鳍鱿)。在组装质量方面, 腹足纲斗嫁
| 纲 | 物种名 | 运行时长/min | 重叠群数目/条 | 基因组覆盖度/% | 基因组准确度/% | 结果文件存储空间占用/GB |
| 双壳纲 | 虾夷蚶蜊 | 1039 | 2 | 100 | 100 | 6.68 |
| 结蚶 | 1119 | 1 | 59.07 | 99.64 | 7.70 | |
| 毛蚶 | 809 | 3 | 43.51 | 26.67 | 7.56 | |
| 多板纲 | 史氏宽板石鳖 | 1067 | 1 | 100 | 100 | 6.08 |
| 红条毛肤石鳖 | 1180 | 1 | 100 | 100 | 8.42 | |
| 琉球花棘石鳖 | 1031 | 1 | 99.60 | 100 | 7.61 | |
| 腹足纲 | 斗嫁 |
1102 | BLAST结果中不包含线粒体基因组序列 | 5.80 | ||
| 三列扭柱螺 | 495 | 1 | 100 | 100 | 4.75 | |
| Pseudosuccinea columella | 2005 | 1 | 0 | 0 | 14.20 | |
| 头足纲 | 菱鳍鱿 | 2292 | 2 | 75.23 | 13.07 | 18.40 |
| 鹦鹉螺 | 2659 | 1 | 100 | 100 | 14.20 | |
| 图氏后乌贼 | 254 | 1 | 59.76 | 100 | 3.43 | |
 |
| 图 5 Ray的基准研究得分图 Fig. 5 Score graph based on the benchmark study of Ray 注: 每个组装软件的得分都是基于组装结果, 并以百分比表示。百分比越高, 组装结果越好 |
在SPAdes组装软体动物主要类群代表物种的组装结果中(表 7, 图 6), SPAdes的运行时长为633 min (斗嫁
| 纲 | 物种名 | 运行时长/min | 重叠群数目/条 | 基因组覆盖度/% | 基因组准确度/% | 结果文件存储空间占用/GB |
| 双壳纲 | 虾夷蚶蜊 | 1565 | 2 | 94.60 | 100 | 23.30 |
| 结蚶 | 1630 | 1 | 58.58 | 100 | 17.80 | |
| 毛蚶 | 699 | 3 | 42.94 | 24.99 | 17.50 | |
| 多板纲 | 史氏宽板石鳖 | 1692 | 1 | 100 | 100 | 17.80 |
| 红条毛肤石鳖 | 1483 | 1 | 100 | 100 | 20.10 | |
| 琉球花棘石鳖 | 924 | 1 | 100 | 100 | 19.10 | |
| 腹足纲 | 斗嫁 |
633 | 8 | 99.52 | 100 | 12.80 |
| 三列扭柱螺 | 2197 | 1 | 39.15 | 100 | 44.30 | |
| Pseudosuccinea columella | 报错并自动退出组装 | |||||
| 头足纲 | 菱鳍鱿 | 报错并自动退出组装 | ||||
| 鹦鹉螺 | 报错并自动退出组装 | |||||
| 图氏后乌贼 | 1423 | 1 | 58.99 | 100 | 6.37 | |
 |
| 图 6 SPAdes的基准研究得分图 Fig. 6 Score graph based on the benchmark study of SPAdes 注: 每个组装软件的得分都是基于组装结果, 并以百分比表示。百分比越高, 组装结果越好 |
在本研究中用到的组装软件中Ray和SPAdes是全基因组组装软件, 并不是专门的线粒体基因组组装软件。Ray是基于指定的k-mer值组装全基因组数据, SPAdes则是在组装时不断调整k-mer以获得最佳的组装效果。MitoZ整合了转录组组装软件SOAPdenovo- Trans (Xie et al, 2014)的思路, 即通过调整k-mer参数对整个基因组进行组装, 然后根据线粒体基因组的平均测序深度远高于核基因组的原理筛选出线粒体Contig(s), 再将测序数据比对到该Contig(s), 不断重复以获得最可靠的组装效果, 然后用隐马尔可夫模型(profile Hidden Markov Model, profile HMM)筛选出可能的线粒体基因组序列, 最后调用Blast、GeneWise、Infernal、MiTFi以及Circos完成线粒体基因组的注释和可视化(Meng et al, 2019)。GetOrganelle提供了embplant_pt、embplant_mt、embplant_nr、fungus_mt、fungus_nr、animal_mt以及other_pt共7种数据库, 在组装时需要根据需求指定数据库, 软件在正式开始组装前将会以此作为“种子”对测序数据中与线粒体基因组相关的reads进行预分群, 从而高效率地获取目标reads并提高线粒体基因组序列的准确性。GetOrganelle在获取相关reads后调用软件SPAdes通过不断调整K-mer进行组装, 与此同时GetOrganelle直接对reads进行错误矫正和错配过滤, 以获得高质量的线粒体基因组全序列(Jin et al, 2020)。MEANGS与GetOrganelle的组装流程有一定的相似性, 都是基于预置的线粒体模块数据库(GetOrganelle: embplant_pt、embplant_mt、embplant_nr、fungus_mt、fungus_nr、animal_mt、other_pt; MEANGS: A-worms、Arthropoda、Bryozoa、Chordata、Echinodermata、Mollusca、Nematoda、N-worms、Porifera-sponges)对测序数据进行预筛选, 高效率地获取目标reads并提高线粒体基因组序列的准确性。但是在后续的组装流程中, GetOrganelle直接调用软件SPAdes通过不断调整K-mer组装这部分reads, 与此同时直接对reads进行错误矫正和错配过滤(Jin et al, 2020)。MEANGS则是根据线性迭代算法(SSAKE)组装预筛选获得的目标reads, 组装获得的线粒体编码重叠群经nhmmer进行二次筛选, 最终得到非冗余线粒体编码重叠群, 在接下来的组装中, 该重叠群作为“种子”序列用于线粒体基因组的组装, 组装结束后MEANGS调用MITOS2对编码基因进行辅助注释(Song et al, 2022)。NOVOPlasty首先将测序数据储存在一个有利于reads快速读取的哈希表(hash table)中, 然后根据使用者提供的线粒体基因组序列片段从测序数据中抓取一条read, 并将这条read反复双向延伸直至获得线粒体基因组序列(Dierckxsens et al, 2017)。NOVOPlasty组装时需要用到的这条参考序列不是直接用于启动线粒体基因组的组装, 而只是用来从基因组浅层测序数据中检索线粒体基因组的read, 以此启动组装流程, 这一步骤有利于减轻完全依赖于参考序列的组装策略中近缘物种的选择问题, 可以接受遗传关系相对较远或较近的物种的线粒体基因组或者序列作为参考序列, 而不会将错配加到组装结果中。
NOVOPlasty是本研究中用到的唯一一个基于参考序列组装策略的线粒体基因组组装软件, 在本研究中运行时间在6~62 min, 结果文件大小均低于0.01 GB, 远低于基于从头组装策略的各个软件。通过比较各个类群的组装结果我们可以看到, NOVOPlasty除在组装双壳纲毛蚶(56 170 bp)时运行失败、在组装头足纲菱鳍鱿时获得的线粒体基因组序列的基因组覆盖度为86.53%, 准确度为77.09%以及在组装头足纲图氏后乌贼时只获得1条基因组覆盖度为61.41%的线粒体基因组序列片段外, 在其他软体动物代表种类的组装中均获得与参考序列近乎完全一致的序列, 更是本研究中唯一一个能够高质量完成结蚶(38 672 bp)的线粒体基因组组装的软件。
基于从头组装策略的组装软件中, MitoZ (57~ 396 min)、GetOrganelle (57~286 min)和MEANGS (54~252 min)的运行时长十分接近, 显著低于全基因组组装软件Ray (254~2 659 min)和SPAdes (633~ 2 197 min); 在结果文件存储空间占用方面, Ray (3.43~18.4 GB)和SPAdes (6.37~44.3 GB)占用最为严重, 其次是MEANGS (3.09~14.9 GB), MitoZ (1.38~ 9.50 GB)和GetOrganelle (0.02~6.77 GB)占用最少。通过比较双壳纲的组装结果我们可以发现, 基于从头组装策略的各个组装软件均能高质量的完成有着常见线粒体基因组大小的虾夷蚶蜊的线粒体基因组的组装, 而在组装线粒体基因组偏大的结蚶(38 672 bp)和毛蚶(56 170 bp)时, GetOrganelle均无法正常运行, MitoZ获得了完整度达83.88%的高质量的结蚶线粒体基因组序列, 但是在组装毛蚶时运行失败, 仅MEANGS以及全基因组组装软件Ray和SPAdes能够成功运行并获得结蚶和毛蚶线粒体基因组的部分序列片段。在多板纲的组装结果中我们可以看到, 各个软件均获得了高质量的线粒体基因组, 仅GetOrganelle和MEANGS在组装琉球花棘石鳖时获得的线粒体基因组序列不完整。在腹足纲P. columella的组装结果中, 仅GetOrganelle和MEANGS能够组装获得高质量的线粒体基因组序列, MitoZ和Ray组装获得的序列基因组覆盖度和准确度均为0%, 而SPAdes无法成功运行。此外, 除Ray在组装斗嫁
本研究对基于不同组装策略的组装软件在软体动物各类群中进行了线粒体基因组组装效果的测试分析。结果表明, 不同的组装软件在软体动物中的组装效果与研究对象的生物学分类无关, 而与线粒体基因组大小有关。基于参考序列组装策略的组装软件, 如本研究中用到的NOVOPlasty在运行时间、结果文件存储空间占用方面表现突出, 远低于从头组装策略的各个软件, 并且组装获得的线粒体基因组序列准确性较高。在基于从头组装策略的组装软件中, 各个软件为了高效地获取高质量的线粒体基因组对组装流程进行了进一步的优化, 但因优化的具体内容不同, 在组装效果方面存在较大的差异, 其中在运行时间、结果文件存储空间占用以及序列质量方面, MEANGS总体优于GetOrganelle和MitoZ, GetOrganelle和MitoZ优于Ray和SPAdes。但是MitoZ实现了其他软件无法实现的“一键式”完成线粒体基因组的获取、注释及可视化工作, Ray和SPAdes能够顺利完成偏大线粒体基因组的组装工作。综上, 基于不同组装策略的组装软件在获取软体动物线粒体基因组时各有优势和不足之处, 在开展组装工作之前, 我们建议根据NCBI中已发表的同科或者同属的物种初步预估研究对象的线粒体基因组的大小, 常见大小的线粒体基因组组装我们建议优先选择NOVOPlasy和MEANGS, 其次是GetOrganelle, 偏大线粒体基因组的组装建议优先选用MEANGS, 其次是Ray和SPAdes。
匡卫民, 于黎, 2019. 基因组时代线粒体基因组拼装策略及软件应用现状. 遗传, 41(11): 979-993 DOI:10.16288/j.yczz.19-227 |
邢晶晶, 2002. 分子遗传标记及其技术在水产生物中的研究与应用. 水产学杂志, 15(1): 61-70 DOI:10.3969/j.issn.1005-3832.2002.01.014 |
李天杰, 曹延祥, 赵红翠, 等, 2016. 动物线粒体基因组测序方法的研究进展. 天津医药, 44(6): 796-800 |
李艳, 黎霞, 陈艳, 2012. 线粒体假基因研究综述. 绵阳师范学院学报, 31(5): 68-75 DOI:10.3969/j.issn.1672-612X.2012.05.017 |
沙淼, 林立亮, 李雪娟, 等, 2013. 线粒体基因组测序策略和方法. 应用昆虫学报, 50(1): 293-297 |
鲁成, 刘运强, 廖顺尧, 等, 2002. 家蚕线粒体基因组全序列测定与分析. 农业生物技术学报, 10(2): 163-170 DOI:10.3969/j.issn.1674-7968.2002.02.015 |
ARNDT A, SMITH M J, 1998. Genetic diversity and population structure in two species of sea cucumber: differing patterns according to mode of development. Molecular Ecology, 7(8): 1053-1064 DOI:10.1046/j.1365-294x.1998.00429.x |
BANKEVICH A, NURK S, ANTIPOV D, et al, 2012. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology, 19(5): 455-477 DOI:10.1089/cmb.2012.0021 |
BERNT M, DONATH A, JüHLING F, et al, 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution, 69(2): 313-319 DOI:10.1016/j.ympev.2012.08.023 |
BOISVERT S, LAVIOLETTE F, CORBEIL J, 2010. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. Journal of Computational Biology, 17(11): 1519-1533 DOI:10.1089/cmb.2009.0238 |
CHENG S, CHANG S Y, GRAVITT P, et al, 1994. Long PCR. Nature, 369(6482): 684-685 DOI:10.1038/369684a0 |
DAVIS C, PETERS D, WARSHAUER D, et al, 2015. Sequencing the hypervariable regions of human mitochondrial DNA using massively parallel sequencing: Enhanced data acquisition for DNA samples encountered in forensic testing. Legal Medicine, 17(2): 123-127 DOI:10.1016/j.legalmed.2014.10.004 |
DIERCKXSENS N, MARDULYN P, SMITS G, 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Research, 45(4): e18 |
DOIRON S, BERNATCHEZ L, BLIER P U, 2002. A comparative mitogenomic analysis of the potential adaptive value of arctic charr mtDNA introgression in brook charr populations (Salvelinus fontinalis Mitchill). Molecular Biology and Evolution, 19(11): 1902-1909 DOI:10.1093/oxfordjournals.molbev.a004014 |
GALASKA P M, LI Y N, KOCOT K M, et al, 2019. Conservation of mitochondrial genome arrangements in brittle stars (Echinodermata, Ophiuroidea). Molecular Phylogenetics and Evolution, 130: 115-120 DOI:10.1016/j.ympev.2018.10.002 |
GHISELLI F, GOMES-DOS-SANTOS A, ADEMA C M, et al, 2021. Molluscan mitochondrial genomes break the rules. Philosophical Transactions of the Royal Society B Biological Sciences, 376(1825): 20200159 DOI:10.1098/rstb.2020.0159 |
GISSI C, IANNELLI F, PESOLE G, 2008. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity, 101(4): 301-320 DOI:10.1038/hdy.2008.62 |
GUREVICH A, SAVELIEV V, VYAHHI N, et al, 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics, 29(8): 1072-1075 DOI:10.1093/bioinformatics/btt086 |
HART A B, SAMUELS D C, HULGAN T, 2013. The other genome: a systematic review of studies of mitochondrial DNA haplogroups and outcomes of HIV infection and antiretroviral therapy. AIDS Reviews, 15(4): 213-220 |
HOFREITER M, SERRE D, POINAR H N, et al, 2001. Ancient DNA. Nature Reviews Genetics, 2(5): 353-359 DOI:10.1038/35072071 |
HUNTER S S, LYON R T, SARVER B A J, et al, 2015. Assembly by Reduced Complexity (ARC): a hybrid approach for targeted assembly of homologous sequences. bioRxiv: 014662 |
JIN J J, YU W B, YANG J B, et al, 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biology, 21(1): 241 DOI:10.1186/s13059-020-02154-5 |
KITANO T, SATO H, TAKAHASHI N, et al, 2022. Complete mitochondrial genomes of three fairy shrimps from snowmelt pools in Japan. BMC Zoology, 7(1): 11 DOI:10.1186/s40850-022-00111-2 |
KONG L F, LI Y N, KOCOT K M, et al, 2020. Mitogenomics reveals phylogenetic relationships of Arcoida (Mollusca, Bivalvia) and multiple independent expansions and contractions in mitochondrial genome size. Molecular Phylogenetics and Evolution, 150: 106857 DOI:10.1016/j.ympev.2020.106857 |
LASLETT D, CANBÄCK B, 2008. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics, 24(2): 172-175 DOI:10.1093/bioinformatics/btm573 |
LI Y N, KOCOT K M, SCHANDER C, et al, 2015. Mitogenomics reveals phylogeny and repeated motifs in control regions of the deep-sea family Siboglinidae (Annelida). Molecular Phylogenetics and Evolution, 85: 221-229 DOI:10.1016/j.ympev.2015.02.008 |
LI H, LIU H Y, SONG F, et al, 2012. Comparative mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS One, 7(9): e45925 DOI:10.1371/journal.pone.0045925 |
LIU Y G, KUROKAWA T, SEKINO M, et al, 2013. Complete mitochondrial DNA sequence of the ark shell Scapharca broughtonii: an ultra-large metazoan mitochondrial genome. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics, 8(1): 72-81 DOI:10.1016/j.cbd.2012.12.003 |
LIU F Y, LI Y L, YU H W, et al, 2021. MolluscDB: an integrated functional and evolutionary genomics database for the hyper-diverse animal phylum Mollusca. Nucleic Acids Research, 49(D1): D988-D997 DOI:10.1093/nar/gkaa918 |
MACHADO D J, LYRA M L, GRANT T, 2016. Mitogenome assembly from genomic multiplex libraries: comparison of strategies and novel mitogenomes for five species of frogs. Molecular Ecology Resources, 16(3): 686-693 DOI:10.1111/1755-0998.12492 |
MENG G L, LI Y Y, YANG C T, et al, 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Research, 47(11): e63 DOI:10.1093/nar/gkz173 |
MIKKELSEN N T, KOCOT K M, HALANYCH K M, 2018. Mitogenomics reveals phylogenetic relationships of caudofoveate aplacophoran molluscs. Molecular Phylogenetics and Evolution, 127: 429-436 DOI:10.1016/j.ympev.2018.04.031 |
MIYA M, KAWAGUCHI A, NISHIDA M, 2001. Mitogenomic exploration of higher teleostean phylogenies: a case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Molecular Biology and Evolution, 18(11): 1993-2009 DOI:10.1093/oxfordjournals.molbev.a003741 |
ORLANDO L, GILBERT M T P, WILLERSLEV E, 2015. Reconstructing ancient genomes and epigenomes. Nature Reviews Genetics, 16(7): 395-408 DOI:10.1038/nrg3935 |
OSIGUS H J, EITEL M, BERNT M, et al, 2013. Mitogenomics at the base of Metazoa. Molecular Phylogenetics and Evolution, 69(2): 339-351 DOI:10.1016/j.ympev.2013.07.016 |
PERSEKE M, BERNHARD D, FRITZSCH G, et al, 2010. Mitochondrial genome evolution in Ophiuroidea, Echinoidea, and Holothuroidea: insights in phylogenetic relationships of Echinodermata. Molecular Phylogenetics and Evolution, 56(1): 201-211 DOI:10.1016/j.ympev.2010.01.035 |
PONDER W F, LINDBERG D R, PONDER J M, 2020. Introducing molluscs[M] // PONDER W F, LINDBERG D R, PONDER J M. Biology and Evolution of the Mollusca. Boca Raton: CRC Press: 1-20.
|
RUBY J G, BELLARE P, DERISI J L, 2013. PRICE: software for the targeted assembly of components of (meta) genomic sequence data. G3 Genes|Genomes|Genetics, 3(5): 865-880 |
SACCONE C, DE GIORGI C, GISSI C, et al, 1999. Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene, 238(1): 195-209 DOI:10.1016/S0378-1119(99)00270-X |
SAIKI R K, SCHARF S, FALOONA F, et al, 1985. Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science, 230(4732): 1350-1354 DOI:10.1126/science.2999980 |
SARMASHGHI S, BOHMANN K, GILBERT M T P, et al, 2019. Skmer: assembly-free and alignment-free sample identification using genome skims. Genome Biology, 20(1): 34 DOI:10.1186/s13059-019-1632-4 |
SMITH D R, SNYDER M, 2007. Complete mitochondrial DNA sequence of the scallop Placopecten magellanicus: Evidence of transposition leading to an uncharacteristically large mitochondrial genome. Journal of Molecular Evolution, 65(4): 380-391 DOI:10.1007/s00239-007-9016-x |
SONG M H, YAN C C, LI J T, 2022. MEANGS: an efficient seed-free tool for de novo assembling animal mitochondrial genome using whole genome NGS data. Briefings in Bioinformatics, 23(1): bbab538 DOI:10.1093/bib/bbab538 |
STRAUB S C K, PARKS M, WEITEMIER K, et al, 2012. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. American Journal of Botany, 99: 349-364 DOI:10.3732/ajb.1100335 |
SUN S E, KONG L F, YU H, et al, 2015. Complete mitochondrial genome of Anadara vellicata (Bivalvia: Arcidae): a unique gene order and large atypical non-coding region. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics, 16: 73-82 DOI:10.1016/j.cbd.2015.08.001 |
SUN S E, LI Q, KONG L F, et al, 2016. Complete mitochondrial genomes of Trisidos kiyoni and Potiarca pilula: varied mitochondrial genome size and highly rearranged gene order in Arcidae. Scientific Reports, 6: 33794 DOI:10.1038/srep33794 |
TAMURA K, AOTSUKA T, 1988. Rapid isolation method of animal mitochondrial DNA by the alkaline lysis procedure. Biochemical Genetics, 26(11/12): 815-819 |
TAMURA K, PETERSON D, PETERSON N, et al, 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28(10): 2731-2739 DOI:10.1093/molbev/msr121 |
THE 1000 GENOMES PROJECT CONSORTIUM, ABECASIS G R, ALTSHULER D, et al, 2010. A map of human genome variation from population-scale sequencing. Nature, 467(7319): 1061-1073 DOI:10.1038/nature09534 |
TZENG C S, HUI C F, SHEN S H, et al, 1992. The complete nucleotide sequence of the Crossostoma lacustre mitochondrial genome: conservation and variations among vertebrates. Nucleic Acids Research, 20(18): 4853-4858 DOI:10.1093/nar/20.18.4853 |
WANG X Y, HUANG Y, LIU N, et al, 2015. Seven complete mitochondrial genome sequences of bushtits (Passeriformes, Aegithalidae, Aegithalos): the evolution pattern in duplicated control regions. Mitochondrial DNA, 26(3): 350-356 DOI:10.3109/19401736.2014.1003821 |
WANG X, ZHANG R G, YUN Q Z, et al, 2021. Comprehensive analysis of complete mitochondrial genome of Sapindus mukorossi Gaertn.: an important industrial oil tree species in China. Industrial Crops and Products, 174: 114210 DOI:10.1016/j.indcrop.2021.114210 |
WEI S J, SHI M, SHARKEY M J, et al, 2010. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics, 11(1): 371 |
WILLIAMS S T, FOSTER P G, HUGHES C, et al, 2017. Curious bivalves: systematic utility and unusual properties of anomalodesmatan mitochondrial genomes. Molecular Phylogenetics and Evolution, 110: 60-72 |
XIE Y L, WU G X, TANG J B, et al, 2014. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics, 30(12): 1660-1666 |
ZHANG H J, ZHANG X, LANDIS J B, et al, 2022. Phylogenomic and comparative analyses of Rheum (Polygonaceae, Polygonoideae). Journal of Systematics and Evolution DOI:10.1111/jse.12814 |
ZHAO H F, CHEN Y, WANG Z T, et al, 2021. Two complete mitogenomes of chalcididae (Hymenoptera: Chalcidoidea): genome description and phylogenetic implications. Insects, 12(12): 1049 |