

瑞氏红鲂 (Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释
(Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释
(Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释
海洋与湖沼  2023, Vol. 54 Issue (5): 1517-1528 2023, Vol. 54 Issue (5): 1517-1528 |

 (Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释
(Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释 (Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释[J]. 海洋与湖沼, 54(5): 1517-1528
(Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释[J]. 海洋与湖沼, 54(5): 1517-1528
 (Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释
(Satyrichthys rieffeli)基因组survey分析及线粒体基因组注释
 (Satyrichthys rieffeli)的基因组特征及线粒体基因组结构, 采用二代高通量测序技术对瑞氏红鲂
(Satyrichthys rieffeli)的基因组特征及线粒体基因组结构, 采用二代高通量测序技术对瑞氏红鲂 进行基因组survey分析, 为研究其分子学内容提供基础信息。利用Illumina nova测序平台进行测序, 组装得到的基因组大小为813 Mb, 杂合率为0.92%, 重复序列比例为45.97%, Contigs的N50大小为712 bp。瑞氏红鲂
进行基因组survey分析, 为研究其分子学内容提供基础信息。利用Illumina nova测序平台进行测序, 组装得到的基因组大小为813 Mb, 杂合率为0.92%, 重复序列比例为45.97%, Contigs的N50大小为712 bp。瑞氏红鲂 线粒体基因组长度为16 527 bp, 共38个基因(22个tRNA、12S rRNA、16S rRNA、ND1~ND6、COⅪ~COXⅢ、Cyt b、ATP8、ATP6、ND4L和1个D-loop区), 基因之间未发生重排, GC含量45.70%; 蛋白质编码基因中出现不完整的密码子T--和TA-; tRNA中只有tRNA-Ser (GCT)缺失了二氢尿苷臂(DHU臂)的简单环, 其他都是正常二级结构。此外, 联合NCBI中18个鲉形目鱼类的线粒体基因组, 利用最大似然法和贝叶斯法构建了鲉形目鱼类的分子系统发育关系。结果表明, 瑞氏红鲂
线粒体基因组长度为16 527 bp, 共38个基因(22个tRNA、12S rRNA、16S rRNA、ND1~ND6、COⅪ~COXⅢ、Cyt b、ATP8、ATP6、ND4L和1个D-loop区), 基因之间未发生重排, GC含量45.70%; 蛋白质编码基因中出现不完整的密码子T--和TA-; tRNA中只有tRNA-Ser (GCT)缺失了二氢尿苷臂(DHU臂)的简单环, 其他都是正常二级结构。此外, 联合NCBI中18个鲉形目鱼类的线粒体基因组, 利用最大似然法和贝叶斯法构建了鲉形目鱼类的分子系统发育关系。结果表明, 瑞氏红鲂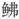 与同为黄鲂
与同为黄鲂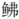 科的须叉吻鲂
科的须叉吻鲂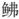 (Scalicus amiscus)为姐妹分支, 支持瑞氏红鲂
(Scalicus amiscus)为姐妹分支, 支持瑞氏红鲂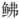 是黄鲂
是黄鲂 科鱼类的形态学分类结果, 为开展黄鲂
科鱼类的形态学分类结果, 为开展黄鲂 科鱼类系统发育深入研究奠定了基础。
科鱼类系统发育深入研究奠定了基础。 基因组survey 线粒体基因组 系统发育关系
基因组survey 线粒体基因组 系统发育关系 高通量测序已成为一种广泛用于动植物基因组测序技术(孟刚等, 2021; 王子寅等, 2023)。通过高通量测序技术可以进行基因组survey研究, 包括基因组大小估计、重复序列比例和GC含量计算, 以及对遗传多样性、基因组结构和遗传改良的了解(Barchi et al, 2011; Rowe et al, 2011; Xu et al, 2014; Shi et al, 2018), 其结果可用于调查基因组图谱和提取线粒体基因组。对于缺乏基因组数据的非模式物种来说, 针对基因组的survey分析是分子机理研究和基因资源开发的前提(Kim et al, 2011)。
线粒体是半自主细胞性细胞器, 普遍存在于真核细胞内(Boore, 1999)。脊椎动物的线粒体DNA (Mitochondrial DNA, mtDNA)基因结构紧凑, 长度一般在15~20 kb之间, 并且编码的基因库非常保守(张方等, 1998)。脊椎动物的线粒体基因组中一般包括了22个tRNA基因、2个rRNA基因、13个蛋白质编码区(PCGs)、1个非编码区包括D-loop区和1个轻链复制起始区(OL) (Satoh et al, 2016)。线粒体基因组具有结构简单、编码区域高度保守、母系遗传、进化速率快、拷贝数高等鲜明特征(Ruan et al, 2020), 被广泛应用于群体遗传学、进化生物学和谱系遗传学等方面(Ko et al, 2018; Zeng et al, 2018; 匡卫民等, 2019; Chan et al, 2019)。线粒体物种特异性DNA片段, 如核糖体RNA (12S rRNA和16S rRNA)、细胞色素b (Cyt b)和细胞色素c氧化酶I (COI)通常用于鱼类物种鉴定(Cutarelli et al, 2018)。目前有较多的学者用线粒体基因组全序列构建系统发育树来比较进化关系, 相比于其他的COI、12S rRNA和16S rRNA等单一基因序列构建的系统发育树可以减少因信息分布的不均一性和序列长度太短而造成的进化树置信度降低带来的影响, 使得研究结果更精确、可靠(肖家光, 2015)。
瑞氏红鲂
本研究所用瑞氏红鲂
用标准的苯酚-氯仿法提取肌肉组织的总基因组DNA (Maniatis et al, 1982)。检测合格的DNA样本通过超声波破碎仪随机打断成长度为300~350 bp的片段, 经末端修复、加A尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备。构建好的文库通过Illumina nova进行PE150测序。使用fastp软件对测序数据进行数据过滤与去冗余操作(Kajitani et al, 2014), 过滤后得到的高质量数据将用于后续的杂合度、GC含量以及基因组大小等信息分析。
1.2.2 基因组大小预估和K-mer分析利用过滤后的数据, 取K=17, 参照Liu等(2013)的K-mer分析方法对瑞氏红鲂
使用SOAPdenovo软件(Luo et al, 2012)对过滤去除低质量数据之后的高质量测序数据进行基因组预组装分析。采用K=48构建Contig和Scaffold, 利用高质量数据进行全基因组装, 使用SOAPdenovo软件将过滤后的reads比对拼接到该组装序列上, 获得原始基因组序列及碱基深度。
1.2.4 线粒体基因组组装和注释借助NOVOPlasty 2.6.3软件从原始的基因组测序数据中提取线粒体基因组序列(Dierckxsens et al, 2017), 参数采用默认设置, 参考序列为GenBank瑞氏红鲂
GenBank中下载18种鲉形目鱼类的线粒体基因组, 以豹鲂
使用Illumina nova测序平台进行双末端测序, 使用瑞氏红鲂
| 名字 | 碱基数 | 碱基大小/Gb | Q20 /% |
Q30 /% |
GC含量/% |
| 原始读数 | 383 941 058 | 57.59 | 96.57 | 91.64 | 41.29 |
| 有效读数 | 329 460 024 | 49.13 | 96.73 | 91.72 | 41.24 |
| 注: Q20: 质量百分比≥20; Q30: 质量百分比≥30 | |||||
为了验证瑞氏红鲂
| 物种 | 比对数量/bp | 比对总数/bp | 占比% | |
| Epinephelus | 石斑鱼属 | 1 185 | 5 945 | 19.93 |
| Cottoperca | 杜父鲈 属 属 |
672 | 5 945 | 11.3 |
| Lateolabrax | 花鲈属 | 549 | 5 945 | 9.23 |
| Plectropomus | 鳃棘鲈属 | 530 | 5 945 | 8.92 |
| Sparus | 鲷属 | 276 | 5 945 | 4.64 |
| Scophthalmus | 菱鲆属 | 267 | 5 945 | 4.49 |
| Sebastes | 平鲉属 | 160 | 5 945 | 2.69 |
| Nibea | 黄姑鱼属 | 144 | 5 945 | 2.42 |
| Anarrhichthys | 狼鳗属 | 134 | 5 945 | 2.25 |
| Trachurus | 竹荚鱼属 | 122 | 5 945 | 2.05 |
| Myripristis | 锯鳞鱼属 | 121 | 5 945 | 2.04 |
| Sphaeramia | 圆竺鲷属 | 120 | 5 945 | 2.02 |
| Larimichthys | 黄鱼属 | 120 | 5 945 | 2.02 |
| Pseudochaenichthys | 拟冰鱼属 | 108 | 5 945 | 1.82 |
| Acanthopagrus | 棘鲷属 | 108 | 5 945 | 1.82 |
| Cyprinus | 鲤属 | 107 | 5 945 | 1.80 |
| Perca | 鲈属 | 100 | 5 945 | 1.68 |
使用K-mer分析预测了瑞氏红鲂
| 样品 | K-mer数 | K-mer深度 | 基因组大小/Mb | 修正后基因组大小/Mb | 杂合率/% | 重复率/% |
| S. rieffeli | 43 850 343 050 | 48 | 827 | 813 | 0.92 | 45.97 |
 |
图 1 瑞氏红鲂 |
通过SOAPdenovo初步组装得到Contigs的结果见表 4。N50和N90的值可以说明survey结果组装的质量, S. rieffeli contig N50和N90分别为712 bp和134 bp, Contigs大于100 bp的占总数的98.42%; A、T、G、C和N的占比分别为29.64%、28.81%、20.52%、21.02%和0, GC含量为41.55%。
| DNA片段 | 总长/bp | 总数(≥1 kb) | 最长/bp | N50/bp | N90/bp | GC含量 | Contigs > 100 bp |
| Contigs | 752 061 430 | 2 083 983 | 31 368 | 712 | 134 | 41.55% | 2 051 006 |
从survey结果里面提取并组装线粒体基因组, 结果显示瑞氏红鲂
 |
图 2 瑞氏红鲂 |
| 基因 | 起始位点 | 终止位点 | 长度/bp | 氨基酸 | 起始密码子 | 终止密码子 | 反密码子 | 基因间隔/bp | 链 |
| tRNA-Phe | 1 | 68 | 68 | GAA | 0 | H | |||
| 12S rRNA | 69 | 1 014 | 946 | 0 | H | ||||
| tRNA-Val | 1 015 | 1 086 | 72 | TAC | 0 | H | |||
| 16S rRNA | 1 087 | 2 785 | 1 699 | 0 | H | ||||
| tRNA-Leu | 2 786 | 2 859 | 74 | TAA | 0 | H | |||
| ND1 | 2 860 | 3 834 | 975 | 324 | ATG | TAG | 0 | H | |
| tRNA-Ile | 3 839 | 3 908 | 70 | GAT | 4 | H | |||
| tRNA-Gln | 3 908 | 3 978 | 71 | TTG | –1 | L | |||
| tRNA-Met | 3 978 | 4 046 | 69 | GAT | –1 | H | |||
| ND2 | 4 047 | 5 092 | 1 046 | 348 | ATG | TA | 0 | H | |
| tRNA-Trp | 5 093 | 5 163 | 71 | TCA | 0 | H | |||
| tRNA-Ala | 5 165 | 5 233 | 69 | TGC | 1 | L | |||
| tRNA-Asn | 5 235 | 5 307 | 73 | GTT | 1 | L | |||
| tRNA-Cys | 5 347 | 5 412 | 66 | GCA | 39 | L | |||
| tRNA-Tyr | 5 413 | 5 482 | 70 | GTA | 0 | L | |||
| COI | 5 484 | 7 034 | 1 551 | 516 | GTG | TAA | 1 | H | |
| tRNA-Ser | 7 035 | 7 105 | 71 | GCT | 0 | L | |||
| tRNA-Asp | 7 109 | 7 181 | 73 | GTC | 3 | H | |||
| COII | 7 189 | 7 879 | 691 | 230 | ATG | T | 7 | H | |
| tRNA-Lys | 7 880 | 7 953 | 74 | TTT | 0 | H | |||
| ATPase 8 | 7 955 | 8 122 | 168 | 55 | ATG | TAA | 1 | H | |
| ATPase 6 | 8 113 | 8 795 | 683 | 227 | ATG | TA | 0 | H | |
| COⅢ | 8 796 | 9 580 | 785 | 261 | ATG | TA | 0 | H | |
| tRNA-Gly | 9 581 | 9 652 | 72 | TCC | 0 | H | |||
| ND3 | 9 653 | 10 001 | 349 | 116 | ATG | T | 0 | H | |
| tRNA-Arg | 10 002 | 10 070 | 69 | TCG | 0 | H | |||
| ND4L | 10 071 | 10 367 | 297 | 98 | ATG | TAA | 0 | H | |
| ND4 | 10 361 | 11 741 | 1 381 | 460 | ATG | T | –5 | H | |
| tRNA-His | 11 742 | 11 810 | 69 | GTG | 0 | H | |||
| tRNA-Ser | 11 811 | 11 878 | 68 | TGA | 0 | H | |||
| tRNA-Leu | 11 884 | 11 956 | 73 | TAG | 5 | H | |||
| ND5 | 11 957 | 13 759 | 1 803 | 600 | ATG | TAA | 0 | H | |
| ND6 | 13 792 | 14 313 | 522 | 173 | ATG | TAG | 32 | L | |
| tRNA-Glu | 14 314 | 14 382 | 69 | TTC | 0 | L | |||
| Cyt b | 14 388 | 15 528 | 1 141 | 380 | ATG | T | 5 | H | |
| tRNA-Thr | 15 529 | 15 600 | 72 | TGT | 0 | H | |||
| tRNA-Pro | 15 600 | 15 669 | 70 | TGG | –1 | L | |||
| D-loop | 15 670 | 16 527 | 858 | 0 | H |
除8个tRNA基因(tRNA-Gln、Ala、Asn、Cys、Tyr、Ser、Glu、Pro)和1个PCGs基因(ND6)在线粒体基因组的L链上, 其他29个基因和D-loop区均在H链上, 基因间都未发生重排。其中11个基因发生了间隔, 共99 bp, tRNA-Cys基因间隔最长, 共39 bp, 其次是基因ND6, 共32 bp; 有4个基因发生了重叠, 共8 bp, 重叠的基因是tRNA-Gln、tRNA-Met、ND4和tRNA-Pro, 其中基因ND4重叠最多, 有5 bp。瑞氏红鲂
| 基因 | T/% | C/% | A/% | G/% | A+T/% | C+G/% | AT-Skew | GC-Skew |
| ND1 | 30.8 | 31 | 22.4 | 15.8 | 53.2 | 46.8 | –0.158 | –0.325 |
| ND2 | 28 | 33.1 | 26.3 | 12.5 | 54.3 | 45.6 | –0.031 | –0.452 |
| COI | 30.5 | 27.3 | 24.2 | 18 | 54.7 | 45.3 | –0.115 | –0.205 |
| COII | 27.4 | 28 | 28.3 | 16.4 | 55.7 | 44.4 | 0.016 | –0.261 |
| ATPase 8 | 21.2 | 36.4 | 30.9 | 11.5 | 52.1 | 47.9 | 0.186 | –0.520 |
| ATPase 6 | 28.6 | 32.9 | 25.3 | 13.2 | 53.9 | 46.1 | –0.061 | –0.427 |
| COⅢ | 28.4 | 29.8 | 24.3 | 17.6 | 52.7 | 47.4 | –0.078 | –0.257 |
| ND3 | 32.8 | 31.3 | 18.7 | 17.2 | 51.5 | 48.5 | –0.274 | –0.291 |
| ND4L | 28.6 | 34.7 | 20.1 | 16.7 | 48.7 | 51.4 | –0.175 | –0.350 |
| ND4 | 28.3 | 30.9 | 25.8 | 14.9 | 54.1 | 45.8 | –0.046 | –0.349 |
| ND5 | 27.8 | 31.0 | 27.0 | 14.1 | 54.8 | 45.1 | –0.015 | –0.375 |
| ND6 | 37.6 | 13.7 | 15.2 | 33.5 | 52.8 | 47.2 | –0.424 | 0.419 |
| Cyt b | 29.1 | 31.8 | 23.8 | 15.3 | 52.9 | 47.1 | –0.100 | –0.350 |
| PCGs | 29.2 | 30.0 | 24.6 | 16.2 | 53.8 | 46.2 | –0.086 | –0.299 |
| tRNA | 26.7 | 21.4 | 27.6 | 24.2 | 54.3 | 45.6 | 0.017 | 0.061 |
| rRNA | 21.9 | 25.3 | 31.9 | 20.9 | 53.8 | 46.2 | 0.186 | –0.095 |
| CR | 30.0 | 21.4 | 32.3 | 16.3 | 62.3 | 37.7 | 0.037 | –0.135 |
| Mitogenome | 26.9 | 28.9 | 27.4 | 16.8 | 54.3 | 45.7 | 0.009 | –0.265 |
瑞氏红鲂
 |
图 3 瑞氏红鲂 |
瑞氏红鲂
 |
图 4 瑞氏红鲂 |
瑞氏红鲂
瑞氏红鲂
 |
图 5 瑞氏红鲂 |
 |
图 6 瑞氏红鲂 |
非编码区又称之为D-环区(displacement-loop region), 位于tRNA-Pro和tRNA-Phe基因之间, 是整个线粒体基因组序列和长度变异最大的区域, 但其中也包含保守片段。瑞氏红鲂
为了分析瑞氏红鲂

 |
图 7 基于瑞氏红鲂 |
 |
图 8 基于瑞氏红鲂 |
近年来, 高通量测序技术的迅速发展为解决非模式海洋鱼类的基因组问题提供了有效的技术手段。利用K-mer方法可以估计非模型物种的基因组大小, 无须任何先验知识(Li et al, 2019b), 其方法已被应用于绒杜父鱼(Hemitripterus villosus) (赵蕊蕊等, 2022)、褐菖鲉(Sebastiscus marmoratus) (Xu et al, 2020)、菖鲉属(Sebastiscus) (Jia et al, 2021)等鲉形目鱼类的研究。目前瑞氏红鲂
本研究组装出来的线粒体基因组大小为16 527 bp, 线粒体结构未出现基因重排现象, GC含量为45.7%, 出现明显的AT偏倚, 其结果符合硬骨鱼类线粒体碱基组成偏好于碱基A和T的特点(Broughton et al, 2001; 蒙子宁等, 2004; Consuegra et al, 2015), 碱基G的含量为16.8%, 表现出明显的反G偏倚(孟刚等, 2021), 与大多数鱼类研究结果一致(丁少雄等, 2006)。13个PCGs中, COI是以GTG作为起始密码子, 其余均为ATG起始密码子, 其中密码子ATG是翻译效率最高的一个(Consuegra et al, 2015)。PCGs终止密码子中出现T--和TA-这类不完整的终止密码子, 是因为这些基因之后是一个编码在同一条链上的基因, 允许转录在没有完整密码子的情况下终止(Hecht et al, 2017)。不完全终止密码子的存在在鱼类的线粒体基因组中很常见, 可以通过在mRNA加工过程中添加一个poly A尾巴来完成(Ojala et al, 1981; Liu et al, 2009)。PCGs的AT-skew和GC-skew为负值(表 6), 一般来说, AT倾斜的幅度小于GC倾斜, 而且在许多情况下, 没有统计学意义, 在这里, AT-skew低于GC-skews (绝对值), 这符合传统的偏好(Yu et al, 2019)。AT-skew的最大值和GC-skew的最小值均出现在ND6中, 且该基因的AT/GC-skew值波动较大, 核苷酸偏斜可能是由于在复制和转录过程中突变压力和选择压力之间的平衡, 为基因复制提供了一个潜在的方向(McLean et al, 1998; Touchon et al, 2008)。tRNA基因结构中仅tRNA-Ser (TCG)基因缺失DHU臂的结构, 这是鱼类线粒体基因组的共同特征, DHU臂的缺失改变了tRNA-Ser (TCG)在线粒体基因中的识别潜力, 使其更容易被识别(Hardt et al, 1993)。瑞氏红鲂
目前通过线粒体基因组构建系统发育树来确定物种的进化关系已经成为系统发育分析的常用方法, 比如周志雄等(2020)通过线粒体基因组构建系统发育树得出东方鲀属内系统发育关系较为复杂, 且存在野生环境下种间自然杂交现象; Miya等(2015)通过过去15年线粒体基因组发展应用做出的总结, 认为线粒体基因组分析和高通量测序技术会成为物种分类重要方法。本研究基于12个PCGs构建系统发育树, 以豹鲂
瑞氏红鲂

丁少雄, 王颖汇, 王军, 等, 2006. 基于16S rDNA部分序列探讨中国近海30种石斑鱼类的分子系统进化关系[J]. 动物学报, 52(3): 504-513. |
王子寅, 刘秉儒, 李子豪, 等, 2023. 基于Survey分析的濒危植物四合木基因组研究[J]. 植物研究, 43(2): 186-193. |
宁子君, 刘玉萍, 张书飞, 等, 2022. 艾氏蛇鳗线粒体基因组全序列结构分析和系统发育关系探讨[J]. 中国水产科学, 29(9): 1264-1276. |
匡卫民, 于黎, 2019. 基因组时代线粒体基因组拼装策略及软件应用现状[J]. 遗传, 41(11): 979-993. |
伍汉霖, 钟俊生, 2021. 中国海洋及河口鱼类系统检索[M]. 北京: 中国农业出版社, 546.
|
肖家光, 2015. 基于线粒体基因组全序列的鱚属鱼类系统发育研究[D]. 青岛: 中国海洋大学: 24-25.
|
张方, 米志勇, 1998. 动物线粒体DNA的分子生物学研究进展[J]. 中国生物工程杂志, 18(3): 25-31, 6. |
陈大刚, 张美昭, 2015. 中国海洋鱼类[M]. 青岛: 中国海洋大学出版社, 811.
|
周丹, 薛仁余, 张晓峰, 等, 2013. 鲤和斑马鱼HOX基因家族同义密码子使用偏性的分析[J]. 水产学杂志, 26(2): 19-25. DOI:10.3969/j.issn.1005-3832.2013.02.004 |
周志雄, 刘波, 宫杰, 等, 2020. 基于线粒体基因组的东方鲀属系统发育学及群体遗传学[J]. 水产学报, 44(11): 1792-1803. |
孟刚, 王瑞娴, 楚渠, 2021. 野桑蚕基因组Survey分析及线粒体基因组注释[J]. 基因组学与应用生物学, 40(S2): 2505-2512. DOI:10.13417/j.gab.040.002505 |
孟乾, 张志勇, 张志伟, 等, 2020. 斑石鲷和条石鲷线粒体基因组密码子使用分析[J]. 水产科学, 39(5): 702-709. DOI:10.16378/j.cnki.1003-1111.2020.05.008 |
赵盛龙, 徐汉祥, 钟俊生, 2016. 浙江海洋鱼类志[M]. 杭州: 浙江科学技术出版社, 523-524.
|
赵蕊蕊, 徐胜勇, 2022. 绒杜父鱼全基因组survey分析及微卫星分布特征[J]. 中国水产科学, 29(7): 994-1001. |
高胜寒, 禹海英, 吴双阳, 等, 2018. 复杂基因组测序技术研究进展[J]. 遗传, 40(11): 944-963. DOI:10.16288/j.yczz.18-255 |
蒙子宁, 庄志猛, 丁少雄, 等, 2004. 中国近海8种石首鱼类的线粒体16S rRNA基因序列变异及其分子系统进化[J]. 自然科学进展, 14(5): 514-521. DOI:10.3321/j.issn:1002-008X.2004.05.006 |
中坊徹次, 2013. 日本産魚類検索全種の同定[M]. 3版. 東海大学出版会: 727-730.
|
BARCHI L, LANTERI S, PORTIS E, et al, 2011. Identification of SNP and SSR markers in eggplant using RAD tag sequencing[J]. BMC Genomics, 12: 304. DOI:10.1186/1471-2164-12-304 |
BOORE J L, 1999. Animal mitochondrial genomes[J]. Nucleic Acids Research, 27(8): 1767-1780. DOI:10.1093/nar/27.8.1767 |
BROUGHTON R E, MILAM J E, ROE B A, 2001. The complete sequence of the zebrafish (Danio rerio) mitochondrial genome and evolutionary patterns in vertebrate mitochondrial DNA[J]. Genome Research, 11(11): 1958-1967. DOI:10.1101/gr.156801 |
BUSSING W A, 2010. A new fish, Peristedion nesium (Scorpaeniformes: Peristediidae) from Isla del Coco, Costa Rica[J]. Revista de Biologia Tropical, 58(4): 1149-1156. |
CHAN E K F, TIMMERMANN A, BALDI B F, et al, 2019. Human origins in a southern African palaeo-wetland and first migrations[J]. Nature, 575(7781): 185-189. DOI:10.1038/s41586-019-1714-1 |
CONSUEGRA S, JOHN E, VERSPOOR E, et al, 2015. Patterns of natural selection acting on the mitochondrial genome of a locally adapted fish species[J]. Genetics Selection Evolution, 47(1): 58. DOI:10.1186/s12711-015-0138-0 |
CUTARELLI A, GALIERO G, CAPUANO F, et al, 2018. Species identification by means of mitochondrial cytochrome b DNA sequencing in processed anchovy, sardine and tuna products[J]. Food and Nutrition Sciences, 9(4): 369-375. DOI:10.4236/fns.2018.94029 |
DIERCKXSENS N, MARDULYN P, SMITS G, 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data[J]. Nucleic Acids Research, 45(4): e18. |
HARDT W D, SCHLEGL J, ERDMANN V A, et al, 1993. Role of the D arm and the anticodon arm in tRNA recognition by eubacterial and eukaryotic RNase P enzymes[J]. Biochemistry, 32(48): 13046-13053. DOI:10.1021/bi00211a014 |
HECHT A, GLASGOW J, JASCHKE P R, et al, 2017. Measurements of translation initiation from all 64 codons in E. coli[J]. Nucleic Acids Research, 45(7): 3615-3626. DOI:10.1093/nar/gkx070 |
IWASAKI W, FUKUNAGA T, ISAGOZAWA R, et al, 2013. MitoFish and mitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline[J]. Molecular Biology and Evolution, 30(11): 2531-2540. DOI:10.1093/molbev/mst141 |
JIA C H, YANG T Y, YANAGIMOTO T, et al, 2021. Comprehensive draft genome analyses of three rockfishes (Scorpaeniformes, Sebastiscus) via genome survey sequencing[J]. Current Issues in Molecular Biology, 43(3): 2048-2058. DOI:10.3390/cimb43030141 |
KAJITANI R, TOSHIMOTO K, NOGUCHI H, et al, 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads[J]. Genome Research, 24(8): 1384-1395. DOI:10.1101/gr.170720.113 |
KAWAI T, 2008. Phylogenetic systematics of the family Peristediidae (Teleostei: Actinopterygii)[J]. Species Diversity, 13(1): 1-34. DOI:10.12782/specdiv.13.1 |
KIM E B, FANG X D, FUSHAN A A, et al, 2011. Genome sequencing reveals insights into physiology and longevity of the naked mole rat[J]. Nature, 479(7372): 223-227. DOI:10.1038/nature10533 |
KO A M S, ZHANG Y Q, YANG M A, et al, 2018. Mitochondrial genome of a 22, 000-year-old giant panda from southern China reveals a new panda lineage[J]. Current Biology, 28(12): R693-R694. DOI:10.1016/j.cub.2018.05.008 |
KUMAR S, STECHER G, LI M, et al, 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms[J]. Molecular Biology and Evolution, 35(6): 1547-1549. DOI:10.1093/molbev/msy096 |
LI Q, WANG Q F, JIN X, et al, 2019a. Characterization and comparison of the mitochondrial genomes from two Lyophyllum fungal species and insights into phylogeny of Agaricomycetes[J]. International Journal of Biological Macromolecules, 121: 364-372. DOI:10.1016/j.ijbiomac.2018.10.037 |
LI Z Y, TIAN C X, HUANG Y, et al, 2019b. A first insight into a draft genome of silver sillago (Sillago sihama) via genome survey sequencing[J]. Animals (Basel), 9(10): 756. |
LIU B H, SHI Y J, YUAN J Y, et al, 2013. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects[J]. arXiv, (1308.2012). |
LIU Y, CUI Z X, 2009. The complete mitochondrial genome sequence of the cutlassfish Trichiurus japonicus (Perciformes: Trichiuridae): Genome characterization and phylogenetic considerations[J]. Marine Genomics, 2(2): 133-142. DOI:10.1016/j.margen.2009.07.003 |
LUO R B, LIU B H, ⅪE Y L, et al, 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler[J]. GigaScience, 1(1): 18. DOI:10.1186/2047-217X-1-18 |
MANIATIS T, FRITSCH E F, SAMBROOK J, 1982. Molecular Cloning[M]. Cold Spring Harbor, USA: Cold Spring Harbor Laboratory.
|
MCLEAN M J, WOLFE K H, DEVINE K M, 1998. Base composition skews, replication orientation, and gene orientation in 12 prokaryote genomes[J]. Journal of Molecular Evolution, 47(6): 691-696. DOI:10.1007/PL00006428 |
MIYA M, NISHIDA M, 2015. The mitogenomic contributions to molecular phylogenetics and evolution of fishes: a 15-year retrospect[J]. Ichthyological Research, 62(1): 29-71. DOI:10.1007/s10228-014-0440-9 |
OJALA D, MONTOYA J, ATTARDI G, 1981. tRNA punctuation model of RNA processing in human mitochondria[J]. Nature, 290(5806): 470-474. DOI:10.1038/290470a0 |
ONO M, KAWAI T, 2014. Review of Armored searobins of the genus Peristedion (Teleostei: Peristediidae) in Japanese waters[J]. Species Diversity, 19(2): 117-131. DOI:10.12782/sd.19.2.117 |
RONQUIST F, HUELSENBECK J P, 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models[J]. Bioinformatics, 19(12): 1572-1574. DOI:10.1093/bioinformatics/btg180 |
ROWE H C, RENAUT S, GUGGISBERG A, 2011. RAD in the realm of next-generation sequencing technologies[J]. Molecular Ecology, 20(17): 3499-3502. |
RUAN H T, LI M, LI Z H, et al, 2020. Comparative analysis of complete mitochondrial genomes of three gerres fishes (Perciformes: Gerreidae) and primary exploration of their evolution history[J]. International Journal of Molecular Sciences, 21(5): 1874. DOI:10.3390/ijms21051874 |
SATOH T P, MIYA M, MABUCHI K, et al, 2016. Structure and variation of the mitochondrial genome of fishes[J]. BMC Genomics, 17(1): 719. DOI:10.1186/s12864-016-3054-y |
SHI L L, YI S K, LI Y H, 2018. Genome survey sequencing of red swamp crayfish Procambarus clarkii[J]. Molecular Biology Reports, 45(5): 799-806. DOI:10.1007/s11033-018-4219-3 |
TOUCHON M, ROCHA E P C, 2008. From GC skews to wavelets: a gentle guide to the analysis of compositional asymmetries in genomic data[J]. Biochimie, 90(4): 648-659. DOI:10.1016/j.biochi.2007.09.015 |
WEI S J, SHI M, CHEN X X, et al, 2010. New views on strand asymmetry in insect mitochondrial genomes[J]. PLoS One, 5(9): e12708. DOI:10.1371/journal.pone.0012708 |
XU P, XU S Z, WU X H, et al, 2014. Population genomic analyses from low-coverage RAD-Seq data: a case study on the non-model cucurbit bottle gourd[J]. The Plant Journal, 77(3): 430-442. DOI:10.1111/tpj.12370 |
XU S Y, ZHANG H, GAO T X, 2020. Comprehensive whole genome survey analyses of male and female brown-spotted flathead fish Platycephalus sp. 1[J]. Genomics, 112(6): 4742-4748. DOI:10.1016/j.ygeno.2020.08.030 |
XU S Y, ZHAO L L, ⅪAO S J, et al, 2019. Whole genome resequencing data for three rockfish species of Sebastes. Scientific Data, 6(1): 97. YU P, ZHOU L, ZHOU X Y, et al, 2019. Unusual AT-skew of Sinorhodeus microlepis mitogenome provides new insights into mitogenome features and phylogenetic implications of bitterling fishes[J]. International Journal of Biological Macromolecules, 129: 339-350. DOI:10.1016/j.ijbiomac.2019.01.200 |
ZENG L, WEN J, FAN S G, et al, 2018. Species identification of fish maw (Porcupinefish) products sold on the market using DNA sequencing of 16S rRNA and COI genes[J]. Food Control, 86: 159-162. DOI:10.1016/j.foodcont.2017.11.031 |
ZHAO X, LIU Y X, DU X Q, et al, 2022. Whole-Genome survey analyses provide a new perspective for the evolutionary biology of shimofuri goby, Tridentiger bifasciatus[J]. Animals (Basel), 12(15): 1914. |